Mitochondrial Genetics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
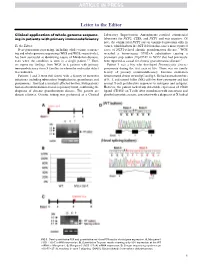
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Two Transgenic Mouse Models for Β-Subunit Components of Succinate-Coa Ligase Yielding Pleiotropic Metabolic Alterations
Two transgenic mouse models for -subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations Kacso, Gergely; Ravasz, Dora; Doczi, Judit; Németh, Beáta; Madgar, Ory; Saada, Ann; Ilin, Polina; Miller, Chaya; Ostergaard, Elsebet; Iordanov, Iordan; Adams, Daniel; Vargedo, Zsuzsanna; Araki, Masatake; Araki, Kimi; Nakahara, Mai; Ito, Haruka; Gál, Aniko; Molnár, Mária J; Nagy, Zsolt; Patocs, Attila; Adam-Vizi, Vera; Chinopoulos, Christos Published in: Biochemical Journal DOI: 10.1042/BCJ20160594 Publication date: 2016 Document version Publisher's PDF, also known as Version of record Document license: CC BY Citation for published version (APA): Kacso, G., Ravasz, D., Doczi, J., Németh, B., Madgar, O., Saada, A., Ilin, P., Miller, C., Ostergaard, E., Iordanov, I., Adams, D., Vargedo, Z., Araki, M., Araki, K., Nakahara, M., Ito, H., Gál, A., Molnár, M. J., Nagy, Z., ... Chinopoulos, C. (2016). Two transgenic mouse models for -subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations. Biochemical Journal, 473(20), 3463-3485. https://doi.org/10.1042/BCJ20160594 Download date: 02. okt.. 2021 Biochemical Journal (2016) 473 3463–3485 DOI: 10.1042/BCJ20160594 Research Article Two transgenic mouse models for β-subunit components of succinate-CoA ligase yielding pleiotropic metabolic alterations Gergely Kacso1,2, Dora Ravasz1,2, Judit Doczi1,2, Beáta Németh1,2, Ory Madgar1,2, Ann Saada3, Polina Ilin3, Chaya Miller3, Elsebet Ostergaard4, Iordan Iordanov1,5, Daniel Adams1,2, Zsuzsanna Vargedo1,2, Masatake -

A Novel Approach to Identify Driver Genes Involved in Androgen-Independent Prostate Cancer
Schinke et al. Molecular Cancer 2014, 13:120 http://www.molecular-cancer.com/content/13/1/120 RESEARCH Open Access A novel approach to identify driver genes involved in androgen-independent prostate cancer Ellyn N Schinke1, Victor Bii1, Arun Nalla1, Dustin T Rae1, Laura Tedrick1, Gary G Meadows1 and Grant D Trobridge1,2* Abstract Background: Insertional mutagenesis screens have been used with great success to identify oncogenes and tumor suppressor genes. Typically, these screens use gammaretroviruses (γRV) or transposons as insertional mutagens. However, insertional mutations from replication-competent γRVs or transposons that occur later during oncogenesis can produce passenger mutations that do not drive cancer progression. Here, we utilized a replication-incompetent lentiviral vector (LV) to perform an insertional mutagenesis screen to identify genes in the progression to androgen-independent prostate cancer (AIPC). Methods: Prostate cancer cells were mutagenized with a LV to enrich for clones with a selective advantage in an androgen-deficient environment provided by a dysregulated gene(s) near the vector integration site. We performed our screen using an in vitro AIPC model and also an in vivo xenotransplant model for AIPC. Our approach identified proviral integration sites utilizing a shuttle vector that allows for rapid rescue of plasmids in E. coli that contain LV long terminal repeat (LTR)-chromosome junctions. This shuttle vector approach does not require PCR amplification and has several advantages over PCR-based techniques. Results: Proviral integrations were enriched near prostate cancer susceptibility loci in cells grown in androgen- deficient medium (p < 0.001), and five candidate genes that influence AIPC were identified; ATPAF1, GCOM1, MEX3D, PTRF, and TRPM4. -

Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma
Anatomy and Pathology Whole-Genome Microarray Detects Deletions and Loss of Heterozygosity of Chromosome 3 Occurring Exclusively in Metastasizing Uveal Melanoma Sarah L. Lake,1 Sarah E. Coupland,1 Azzam F. G. Taktak,2 and Bertil E. Damato3 PURPOSE. To detect deletions and loss of heterozygosity of disease is fatal in 92% of patients within 2 years of diagnosis. chromosome 3 in a rare subset of fatal, disomy 3 uveal mela- Clinical and histopathologic risk factors for UM metastasis noma (UM), undetectable by fluorescence in situ hybridization include large basal tumor diameter (LBD), ciliary body involve- (FISH). ment, epithelioid cytomorphology, extracellular matrix peri- ϩ ETHODS odic acid-Schiff-positive (PAS ) loops, and high mitotic M . Multiplex ligation-dependent probe amplification 3,4 5 (MLPA) with the P027 UM assay was performed on formalin- count. Prescher et al. showed that a nonrandom genetic fixed, paraffin-embedded (FFPE) whole tumor sections from 19 change, monosomy 3, correlates strongly with metastatic death, and the correlation has since been confirmed by several disomy 3 metastasizing UMs. Whole-genome microarray analy- 3,6–10 ses using a single-nucleotide polymorphism microarray (aSNP) groups. Consequently, fluorescence in situ hybridization were performed on frozen tissue samples from four fatal dis- (FISH) detection of chromosome 3 using a centromeric probe omy 3 metastasizing UMs and three disomy 3 tumors with Ͼ5 became routine practice for UM prognostication; however, 5% years’ metastasis-free survival. to 20% of disomy 3 UM patients unexpectedly develop metas- tases.11 Attempts have therefore been made to identify the RESULTS. Two metastasizing UMs that had been classified as minimal region(s) of deletion on chromosome 3.12–15 Despite disomy 3 by FISH analysis of a small tumor sample were found these studies, little progress has been made in defining the key on MLPA analysis to show monosomy 3. -
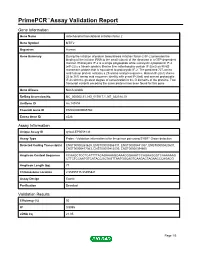
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mitochondrial translational initiation factor 2 Gene Symbol MTIF2 Organism Human Gene Summary During the initiation of protein biosynthesis initiation factor-2 (IF-2) promotes the binding of the initiator tRNA to the small subunit of the ribosome in a GTP-dependent manner. Prokaryotic IF-2 is a single polypeptide while eukaryotic cytoplasmic IF-2 (eIF-2) is a trimeric protein. Bovine liver mitochondria contain IF-2(mt) an 85-kD monomeric protein that is equivalent to prokaryotic IF-2. The predicted 727-amino acid human protein contains a 29-amino acid presequence. Human IF-2(mt) shares 32 to 38% amino acid sequence identity with yeast IF-2(mt) and several prokaryotic IF-2s with the greatest degree of conservation in the G domains of the proteins. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases Not Available RefSeq Accession No. NC_000002.11, NG_017017.1, NT_022184.15 UniGene ID Hs.149894 Ensembl Gene ID ENSG00000085760 Entrez Gene ID 4528 Assay Information Unique Assay ID qHsaCEP0058136 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000263629, ENST00000366137, ENST00000441307, ENST00000420637, ENST00000417363, ENST00000412530, ENST00000394600 Amplicon Context Sequence CCAAGCTCCTCATTTTACAGAAAAGGAAACGGAAATCCAGAAGGGTCAAAAAAG CTTCTCCAATGTCATACCGCTAGTTAATGGCAGTCAAGACTAGAACCCAGACG Amplicon Length (bp) 77 Chromosome Location 2:55495715-55495821 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9995 cDNA Cq 21.05 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.56 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Paraganglioma (PGL) Tumors in Patients with Succinate Dehydrogenase-Related PCC–PGL Syndromes: a Clinicopathological and Molecular Analysis
T G Papathomas and others Non-PCC/PGL tumors in the SDH 170:1 1–12 Clinical Study deficiency Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC–PGL syndromes: a clinicopathological and molecular analysis Thomas G Papathomas1, Jose Gaal1, Eleonora P M Corssmit2, Lindsey Oudijk1, Esther Korpershoek1, Ketil Heimdal3, Jean-Pierre Bayley4, Hans Morreau5, Marieke van Dooren6, Konstantinos Papaspyrou7, Thomas Schreiner8, Torsten Hansen9, Per Arne Andresen10, David F Restuccia1, Ingrid van Kessel6, Geert J L H van Leenders1, Johan M Kros1, Leendert H J Looijenga1, Leo J Hofland11, Wolf Mann7, Francien H van Nederveen12, Ozgur Mete13,14, Sylvia L Asa13,14, Ronald R de Krijger1,15 and Winand N M Dinjens1 1Department of Pathology, Josephine Nefkens Institute, Erasmus MC, University Medical Center, PO Box 2040, 3000 CA Rotterdam, The Netherlands, 2Department of Endocrinology, Leiden University Medical Center, Leiden,The Netherlands, 3Section for Clinical Genetics, Department of Medical Genetics, Oslo University Hospital, Oslo, Norway, 4Department of Human and Clinical Genetics, Leiden University Medical Center, Leiden, The Netherlands, 5Department of Pathology, Leiden University Medical Center, Leiden, The Netherlands, 6Department of Clinical Genetics, Erasmus MC, University Medical Center, Rotterdam, The Netherlands, 7Department of Otorhinolaryngology, Head and Neck Surgery, University Medical Center of the Johannes Gutenberg University Mainz, Mainz, Germany, 8Section for Specialized Endocrinology, -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Primate Specific Retrotransposons, Svas, in the Evolution of Networks That Alter Brain Function
Title: Primate specific retrotransposons, SVAs, in the evolution of networks that alter brain function. Olga Vasieva1*, Sultan Cetiner1, Abigail Savage2, Gerald G. Schumann3, Vivien J Bubb2, John P Quinn2*, 1 Institute of Integrative Biology, University of Liverpool, Liverpool, L69 7ZB, U.K 2 Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK 3 Division of Medical Biotechnology, Paul-Ehrlich-Institut, Langen, D-63225 Germany *. Corresponding author Olga Vasieva: Institute of Integrative Biology, Department of Comparative genomics, University of Liverpool, Liverpool, L69 7ZB, [email protected] ; Tel: (+44) 151 795 4456; FAX:(+44) 151 795 4406 John Quinn: Department of Molecular and Clinical Pharmacology, Institute of Translational Medicine, The University of Liverpool, Liverpool L69 3BX, UK, [email protected]; Tel: (+44) 151 794 5498. Key words: SVA, trans-mobilisation, behaviour, brain, evolution, psychiatric disorders 1 Abstract The hominid-specific non-LTR retrotransposon termed SINE–VNTR–Alu (SVA) is the youngest of the transposable elements in the human genome. The propagation of the most ancient SVA type A took place about 13.5 Myrs ago, and the youngest SVA types appeared in the human genome after the chimpanzee divergence. Functional enrichment analysis of genes associated with SVA insertions demonstrated their strong link to multiple ontological categories attributed to brain function and the disorders. SVA types that expanded their presence in the human genome at different stages of hominoid life history were also associated with progressively evolving behavioural features that indicated a potential impact of SVA propagation on a cognitive ability of a modern human. -

Genetics of Amyotrophic Lateral Sclerosis in the Han Chinese
Genetics of amyotrophic lateral sclerosis in the Han Chinese Ji He A thesis submitted for the degree of Master of Philosophy at The University of Queensland in 2015 The University of Queensland Diamantina Institute 1 Abstract Amyotrophic lateral sclerosis is the most frequently occurring neuromuscular degenerative disorders, and has an obscure aetiology. Whilst major progress has been made, the majority of the genetic variation involved in ALS is, as yet, undefined. In this thesis, multiple genetic studies have been conducted to advance our understanding of the genetic architecture of the disease. In the light of the paucity of comprehensive genetic studies performed in Chinese, the presented study focused on advancing our current understanding in genetics of ALS in the Han Chinese population. To identify genetic variants altering risk of ALS, a genome-wide association study (GWAS) was performed. The study included 1,324 Chinese ALS cases and 3,115 controls. After quality control, a number of analyses were performed in a cleaned dataset of 1,243 cases and 2,854 controls that included: a genome-wide association analysis to identify SNPs associated with ALS; a genomic restricted maximum likelihood (GREML) analysis to estimate the proportion of the phenotypic variance in ALS liability due to common SNPs; and a gene- based analysis to identify genes associated with ALS. There were no genome-wide significant SNPs or genes associated with ALS. However, it was estimated that 17% (SE: 0.05; P=6×10-5) of the phenotypic variance in ALS liability was due to common SNPs. The top associated SNP was within GNAS (rs4812037; p =7×10-7). -

Timing of Antioxidant Gene Therapy: Implications for Treating Dry AMD
Biochemistry and Molecular Biology Timing of Antioxidant Gene Therapy: Implications for Treating Dry AMD Manas R. Biswal,1 Pingyang Han,1 Ping Zhu,2 Zhaoyang Wang,3 Hong Li,1 Cristhian J. Ildefonso,2 and Alfred S. Lewin1 1Department of Molecular Genetics and Microbiology, University of Florida College of Medicine, Gainesville, Florida, United States 2Department of Ophthalmology, University of Florida College of Medicine, Gainesville, Florida, United States 3Department of Ophthalmology, Shanghai Ninth People’s Hospital, Shanghai Jiaotong University School of Medicine, Huangpu District, Shanghai, China Correspondence: Manas R. Biswal, PURPOSE. To investigate whether antioxidant gene therapy protects the structure and function Department of Molecular Genetics of retina in a murine model of RPE atrophy, and to determine whether antioxidant gene and Microbiology, University of Flor- therapy can prevent degeneration once it has begun. ida College of Medicine, 1200 New- ell Drive, Gainesville, FL 32610, USA; METHODS. We induced mitochondrial oxidative stress in RPE by conditional deletion of Sod2, Biswal@ufl.edu. the gene for manganese superoxide dismutase (MnSOD). These mice exhibited localized Submitted: December 9, 2016 atrophy of the RPE and overlying photoreceptors. We restored Sod2 to the RPE of one eye Accepted: January 23, 2017 using adeno-associated virus (AAV) by subretinal injection at an early (6 weeks) and a late Citation: Biswal MR, Han P, Zhu P, et stage (6 months), injecting the other eye with an AAV vector expressing green fluorescent al. Timing of antioxidant gene thera- protein (GFP). Retinal degeneration was monitored over a period of 9 months by py: implications for treating dry AMD. electroretinography (ERG) and spectral-domain optical coherence tomography (SD-OCT). -

1 UST College of Science Department of Biological Sciences
UST College of Science Department of Biological Sciences 1 Pharmacogenomics of Myofascial Pain Syndrome An Undergraduate Thesis Submitted to the Department of Biological Sciences College of Science University of Santo Tomas In Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in Biology Jose Marie V. Lazaga Marc Llandro C. Fernandez May 2021 UST College of Science Department of Biological Sciences 2 PANEL APPROVAL SHEET This undergraduate research manuscript entitled: Pharmacogenomics of Myofascial Pain Syndrome prepared and submitted by Jose Marie V. Lazaga and Marc Llandro C. Fernandez, was checked and has complied with the revisions and suggestions requested by panel members after thorough evaluation. This final version of the manuscript is hereby approved and accepted for submission in partial fulfillment of the requirements for the degree of Bachelor of Science in Biology. Noted by: Asst. Prof. Marilyn G. Rimando, PhD Research adviser, Bio/MicroSem 602-603 Approved by: Bio/MicroSem 603 panel member Bio/MicroSem 603 panel member Date: Date: UST College of Science Department of Biological Sciences 3 DECLARATION OF ORIGINALITY We hereby affirm that this submission is our own work and that, to the best of our knowledge and belief, it contains no material previously published or written by another person nor material to which a substantial extent has been accepted for award of any other degree or diploma of a university or other institute of higher learning, except where due acknowledgement is made in the text. We also declare that the intellectual content of this undergraduate research is the product of our work, even though we may have received assistance from others on style, presentation, and language expression.