Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome Sequence of the Progenitor of the Wheat D Genome Aegilops Tauschii Ming-Cheng Luo1*, Yong Q
OPEN LETTER doi:10.1038/nature24486 Genome sequence of the progenitor of the wheat D genome Aegilops tauschii Ming-Cheng Luo1*, Yong Q. Gu2*, Daniela Puiu3*, Hao Wang4,5,6*, Sven O. Twardziok7*, Karin R. Deal1, Naxin Huo1,2, Tingting Zhu1, Le Wang1, Yi Wang1,2, Patrick E. McGuire1, Shuyang Liu1, Hai Long1, Ramesh K. Ramasamy1, Juan C. Rodriguez1, Sonny L. Van1, Luxia Yuan1, Zhenzhong Wang1,8, Zhiqiang Xia1, Lichan Xiao1, Olin D. Anderson2, Shuhong Ouyang2,8, Yong Liang2,8, Aleksey V. Zimin3, Geo Pertea3, Peng Qi4,5, Jeffrey L. Bennetzen6, Xiongtao Dai9, Matthew W. Dawson9, Hans-Georg Müller9, Karl Kugler7, Lorena Rivarola-Duarte7, Manuel Spannagl7, Klaus F. X. Mayer7,10, Fu-Hao Lu11, Michael W. Bevan11, Philippe Leroy12, Pingchuan Li13, Frank M. You13, Qixin Sun8, Zhiyong Liu8, Eric Lyons14, Thomas Wicker15, Steven L. Salzberg3,16, Katrien M. Devos4,5 & Jan Dvořák1 Aegilops tauschii is the diploid progenitor of the D genome of We conclude therefore that the size of the Ae. tauschii genome is about hexaploid wheat1 (Triticum aestivum, genomes AABBDD) and 4.3 Gb. an important genetic resource for wheat2–4. The large size and To assess the accuracy of our assembly, sequences of 195 inde- highly repetitive nature of the Ae. tauschii genome has until now pendently sequenced and assembled AL8/78 BAC clones8, which precluded the development of a reference-quality genome sequence5. contained 25,540,177 bp in 2,405 unordered contigs, were aligned to Here we use an array of advanced technologies, including ordered- Aet v3.0. Five contigs failed to align and six extended partly into gaps, clone genome sequencing, whole-genome shotgun sequencing, accounting for 0.25% of the total length of the contigs. -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -

Bioinformatics Integrated Analysis to Investigate Candidate Biomarkers and Associated Metabolites in Osteosarcoma
Wang et al. Journal of Orthopaedic Surgery and Research (2021) 16:432 https://doi.org/10.1186/s13018-021-02578-0 RESEARCH ARTICLE Open Access Bioinformatics integrated analysis to investigate candidate biomarkers and associated metabolites in osteosarcoma Jun Wang, Mingzhi Gong, Zhenggang Xiong, Yangyang Zhao and Deguo Xing* Abstract Background: This study hoped to explore the potential biomarkers and associated metabolites during osteosarcoma (OS) progression based on bioinformatics integrated analysis. Methods: Gene expression profiles of GSE28424, including 19 human OS cell lines (OS group) and 4 human normal long bone tissue samples (control group), were downloaded. The differentially expressed genes (DEGs) in OS vs. control were investigated. The enrichment investigation was performed based on DEGs, followed by protein–protein interaction network analysis. Then, the feature genes associated with OS were explored, followed by survival analysis to reveal prognostic genes. The qRT-PCR assay was performed to test the expression of these genes. Finally, the OS- associated metabolites and disease-metabolic network were further investigated. Results: Totally, 357 DEGs were revealed between the OS vs. control groups. These DEGs, such as CXCL12, were mainly involved in functions like leukocyte migration. Then, totally, 38 feature genes were explored, of which 8 genes showed significant associations with the survival of patients. High expression of CXCL12, CEBPA, SPARCL1, CAT, TUBA1A, and ALDH1A1 was associated with longer survival time, while high expression of CFLAR and STC2 was associated with poor survival. Finally, a disease-metabolic network was constructed with 25 nodes including two disease-associated metabolites cyclophosphamide and bisphenol A (BPA). BPA showed interactions with multiple prognosis-related genes, such as CXCL12 and STC2. -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -
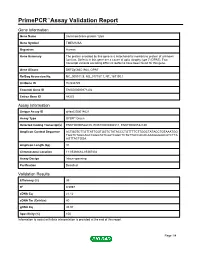
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name transmembrane protein 126A Gene Symbol TMEM126A Organism Human Gene Summary The protein encoded by this gene is a mitochondrial membrane protein of unknown function. Defects in this gene are a cause of optic atrophy type 7 (OPA7). Two transcript variants encoding different isoforms have been found for this gene. Gene Aliases DKFZp586C1924, OPA7 RefSeq Accession No. NC_000011.9, NG_017157.1, NT_167190.1 UniGene ID Hs.533725 Ensembl Gene ID ENSG00000171202 Entrez Gene ID 84233 Assay Information Unique Assay ID qHsaCID0014621 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000528105, ENST00000304511, ENST00000532180 Amplicon Context Sequence ACTGGTCTTGTTATTGGTGGTCTATACCCTGTTTTCTTGGCTATACCTGTAAATGG TGGTCTAGCAGCCAGGTATCAATCAGCTCTGTTACCACACAAAGGGAACATCTTA AGTTACTGGA Amplicon Length (bp) 91 Chromosome Location 11:85366682-85367402 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 99 R2 0.9997 cDNA Cq 21.12 cDNA Tm (Celsius) 80 gDNA Cq 35.93 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report TMEM126A, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells
Original Endocrinol Metab 2016;31:336-342 http://dx.doi.org/10.3803/EnM.2016.31.2.336 Article pISSN 2093-596X · eISSN 2093-5978 The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells So Young Park1, Sung Hoon Kim1, Hyun Koo Yoon1, Chang Hoon Yim1, Sung-Kil Lim2 1Department of Internal Medicine, Cheil General Hospital & Women’s Healthcare Center, Dankook University College of Medicine, Seoul; 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea Background: Reactive oxygen species (ROS) and antioxidants are associated with maintenance of cellular function and metabo- lism. Nuclear factor-E2-related factor 1 (NFE2L1, Nrf1) is known to regulate the expression of a number of genes involved in oxidative stress and inflammation. The purpose of this study was to examine the effects of NFE2L1 on the response to oxidative stress in osteoblastic MC3T3-E1 cells. Methods: The murine calvaria-derived MC3T3-E1 cell line was exposed to lipopolysaccharide (LPS) for oxidative stress induc- tion. NFE2L1 effects were evaluated using small interfering RNA (siRNA) for NFE2L1 mRNA. ROS generation and the levels of known antioxidant enzyme genes were assayed. Results: NFE2L1 expression was significantly increased 2.4-fold compared to the control group at 10 μg/mL LPS in MC3T3-E1 cells (P<0.05). LPS increased formation of intracellular ROS in MC3T3-E1 cells. NFE2L1 knockdown led to an additional in- crease of ROS (20%) in the group transfected with NFE2L1 siRNA compared with the control group under LPS stimulation (P<0.05). -

Os9) and the Hypoxia Response Gene, Hypoxia Inducible Factor 1 (Hif1) Alpha, in Kidney Tubular Cells
North Carolina Agricultural and Technical State University Aggie Digital Collections and Scholarship Theses Electronic Theses and Dissertations 2013 Meprin Interaction With Osteosarcoma 9 (Os9) And The Hypoxia Response Gene, Hypoxia Inducible Factor 1 (Hif1) Alpha, In Kidney Tubular Cells Barry L. Martin North Carolina Agricultural and Technical State University Follow this and additional works at: https://digital.library.ncat.edu/theses Recommended Citation Martin, Barry L., "Meprin Interaction With Osteosarcoma 9 (Os9) And The Hypoxia Response Gene, Hypoxia Inducible Factor 1 (Hif1) Alpha, In Kidney Tubular Cells" (2013). Theses. 287. https://digital.library.ncat.edu/theses/287 This Thesis is brought to you for free and open access by the Electronic Theses and Dissertations at Aggie Digital Collections and Scholarship. It has been accepted for inclusion in Theses by an authorized administrator of Aggie Digital Collections and Scholarship. For more information, please contact [email protected]. Meprin Interaction with Osteosarcoma 9 (OS9) and the Hypoxia Response Gene, Hypoxia- inducible Factor 1 (HIF1) Alpha, in Kidney Tubular Cells Barry L. Martin North Carolina A&T State University A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Department: Biology Major: Biology Major Professor: Dr. Elimelda Moige Ongeri Greensboro, North Carolina 2013 i School of Graduate Studies North Carolina Agricultural and Technical State University This is to certify that the Master’s Thesis of Barry L. Martin has met the thesis requirements of North Carolina Agricultural and Technical State University Greensboro, North Carolina 2013 Approved by: Elimelda Moige Ongeri, PhD Jian Han, PhD Major Professor Committee Member Catherine White, PhD Scott Harrison, PhD Committee Member Committee Member Mary Smith, PhD Sanjiv Sarin, PhD Department Chair Dean, The Graduate School ii © Copyright by Barry L. -

A Genome-Wide Association Study
ISSN (Online) 2287-3406 Journal of Life Science 2021 Vol. 31. No. 6. 568~573 DOI : https://doi.org/10.5352/JLS.2021.31.6.568 - Note - The Association of Long Noncoding RNA LOC105372577 with Endoplasmic Reticulum Protein 29 Expression: A Genome-wide Association Study Soyeon Lee1, Kiang Kwon2, Younghwa Ko3 and O-Yu Kwon3* 1School of Systems Biomedical Science, College of Natural Sciences, Soongsil University, Seoul 06978, Korea 2Department of Clinical Laboratory Science, Wonkwang Health Science University, Iksan 54538, Korea 3Department of Anatomy & Cell Biology, College of Medicine, Chungnam National University, Daejeon 35015, Korea Received April 27, 2021 /Revised April 29, 2021 /Accepted April 30, 2021 This study identified genomic factors associated with endoplasmic reticulum protein (ERp)29 gene ex- pression in a genome-wide association study (GWAS) of genetic variants, including single-nucleotide polymorphisms (SNPs). In total, 373 European genes from the 1000 Genomes Project were analyzed. SNPs with an allelic frequency of less than or more than 5% were removed, resulting in 5,913,563 SNPs including in the analysis. The following expression quantitative trait loci (eQTL) from the long noncoding RNA LOC105372577 were strongly associated with ERp29 expression: rs6138266 (p<4.172e10), rs62193420 (p<1.173e10), and rs6138267 (p<2.041e10). These were strongly expressed in the testis and in the brain. The three eQTL were identified through a transcriptome-wide association study (TWAS) and showed a significant association with ERp29 and osteosarcoma amplified 9 (OS9) expression. Upstream sequences of rs6138266 were recognized by ChIP-seq data, while HaploReg was used to demonstrate how its regulatory DNA binds upstream of transcription factor 1 (USF1). -

University of Cincinnati
UNIVERSITY OF CINCINNATI _____________ , 20 _____ I,______________________________________________, hereby submit this as part of the requirements for the degree of: ________________________________________________ in: ________________________________________________ It is entitled: ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ Approved by: ________________________ ________________________ ________________________ ________________________ ________________________ Role of Glutamate-Cysteine Ligase in Maintaining Glutathione Homeostasis and Protecting against Oxidative Stress A dissertation submitted to the Division of Research and Advanced Studies University of Cincinnati in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (Ph.D.) in the Department of Environmental Health of the College of Medicine 2003 by Yi Yang M.D., Sun Yat-sen University of Medical Sciences, 1995 M.S., Sun Yat-sen University of Medical Sciences, 1997 Committee Chair: Daniel W. Nebert, M.D. Professor Department of Environmental Health University of Cincinnati Abstract Glutamate-cysteine ligase (GCL) is the rate-limiting enzyme catalyzing the first step of glutathione (GSH) biosynthesis. In higher eukaryotes, this enzyme is a heterodimer comprising a catalytic subunit (GCLC) and a modifier subunit (GCLM); the latter changes the catalytic characteristics of the holoenzyme. In the first part -

Distinct Isoforms of Nrf1 Diversely Regulate Different Subsets of Its Cognate Target Genes
bioRxiv preprint doi: https://doi.org/10.1101/356071; this version posted June 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Distinct isoforms of Nrf1 diversely regulate different subsets of its cognate target genes Meng Wang, Lu Qiu, Xufang Ru, Yijiang Song, Yiguo Zhang* The Laboratory of Cell Biochemistry and Topogenetic Regulation, College of Bioengineering and Faculty of Sciences, Chongqing University, No. 174 Shazheng Street, Shapingba District, Chongqing 400044, China *Correspondence should be addressed to Yiguo Zhang (email: [email protected] or [email protected]). Abstract The single Nrf1 gene has capability to be differentially transcripted alongside with alternative mRNA-splicing and subsequent translation through different initiation signals so as to yield distinct lengths of polypeptide isoforms. Amongst them, three of the most representatives are Nrf1α, Nrf1β and Nrf1γ, but the putative specific contribution of each isoform to regulating ARE-driven target genes remains unknown. To address this, we have here established three cell lines on the base of the Flp-In™ T-REx™ system, which are allowed for tetracycline-inducibly stable expression of Nrf1α, Nrf1β and Nrf1γ. The RNA-Sequencing results have demonstrated that a vast majority of differentially expressed genes (i.e. 90 DEGs detected) were dominantly up-regulated by Nrf1α and/or Nrf1β following induction by tetracycline. By contrast, other DEGs regulated by Nrf1γ were far less than those regulated by Nrf1α/β (i.e. ~11 of Nrf1α and 7 of Nrf1β). Further transcriptomic analysis revealed that tetracycline-induced expression of Nrf1γ significantly increased the percentage of down-regulated genes in total DEGs. -
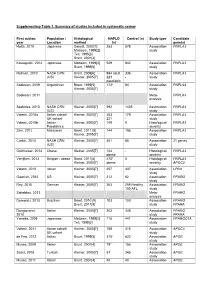
Supplementary Table 1. Summary of Studies Included in Systematic Review
Supplementary Table 1. Summary of studies included in systematic review First author, Population / Histological NAFLD Control (n) Study type Candidate year Location method (n) gene(s) Hotta, 2010 Japanese Sanyal, 2002[1] 253 578 Association PNPLA3 Matteoni, 1999[2] study Teli, 1995[3] Brunt, 2001[4] Kawaguchi, 2012 Japanese Matteoni, 1999[2] 529 942 Association PNPLA3 Brunt, 1999[5] study Rotman, 2010 NASH CRN Brunt, 2009[6] 894 adult 336 Association PNPLA3 (US) Kleiner, 2005[7] 223 - study paediatric Sookoian, 2009 Argentinian Brunt, 1999[5] 172* 94 Association PNPLA3 Kleiner, 2005[7] study Sookoian, 2011 Meta- PNPLA3 analysis Speliotes, 2010 NASH CRN Kleiner, 2005[7] 592 1405 Association PNPLA3 (US) study Valenti, 2010a Italian cohort/ Kleiner, 2005[7] 253 179 Association PNPLA3 UK cohort 321 - study Valenti, 2010b Italian - Kleiner, 2005[7] 149 0 Histological PNPLA3 Paediatrics severity Zain, 2012 Malaysian Brunt, 2011 [8] 144 198 Association PNPLA3 Kleiner, 2005[7] study Corbin, 2013 NASH CRN Kleiner, 2005[7] 361 85 Association 21 genes (US) study Guichelaar, 2013 Obese Kleiner, 2005[7] 144 - Histological PNPLA3 obese severity Verrijken, 2013 Belgian - obese Brunt, 2011[8] 470* 0 Histological PNPLA3 Kleiner, 2005[7] obese severity APOC3 Valenti, 2012 Italian Kleiner, 2005[7] 257 337 Association LPIN1 study Gawrieh, 2012 US Kleiner, 2005[7] 212 62 Association PPARG study Rey, 2010 German Kleiner, 2005[7] 263 259 Healthy Association PPARG 100 AFL study Sahebkar, 2013 Meta- PPARG analysis Domenici, 2013 Brazilian Brunt, 2010 [9] 103 103 -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors.