University of Cincinnati
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

High-Throughput Bioinformatics Approaches to Understand Gene Expression Regulation in Head and Neck Tumors
High-throughput bioinformatics approaches to understand gene expression regulation in head and neck tumors by Yanxiao Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2016 Doctoral Committee: Associate Professor Maureen A. Sartor, Chair Professor Thomas E. Carey Assistant Professor Hui Jiang Professor Ronald J. Koenig Associate Professor Laura M. Rozek Professor Kerby A. Shedden c Yanxiao Zhang 2016 All Rights Reserved I dedicate this thesis to my family. For their unfailing love, understanding and support. ii ACKNOWLEDGEMENTS I would like to express my gratitude to Dr. Maureen Sartor for her guidance in my research and career development. She is a great mentor. She patiently taught me when I started new in this field, granted me freedom to explore and helped me out when I got lost. Her dedication to work, enthusiasm in teaching, mentoring and communicating science have inspired me to feel the excite- ment of research beyond novel scientific discoveries. I’m also grateful to have an interdisciplinary committee. Their feedback on my research progress and presentation skills is very valuable. In particular, I would like to thank Dr. Thomas Carey and Dr. Laura Rozek for insightful discussions on the biology of head and neck cancers and human papillomavirus, Dr. Ronald Koenig for expert knowledge on thyroid cancers, Dr. Hui Jiang and Dr. Kerby Shedden for feedback on the statistics part of my thesis. I would like to thank all the past and current members of Sartor lab for making the lab such a lovely place to stay and work in. -

The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells
Original Endocrinol Metab 2016;31:336-342 http://dx.doi.org/10.3803/EnM.2016.31.2.336 Article pISSN 2093-596X · eISSN 2093-5978 The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells So Young Park1, Sung Hoon Kim1, Hyun Koo Yoon1, Chang Hoon Yim1, Sung-Kil Lim2 1Department of Internal Medicine, Cheil General Hospital & Women’s Healthcare Center, Dankook University College of Medicine, Seoul; 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea Background: Reactive oxygen species (ROS) and antioxidants are associated with maintenance of cellular function and metabo- lism. Nuclear factor-E2-related factor 1 (NFE2L1, Nrf1) is known to regulate the expression of a number of genes involved in oxidative stress and inflammation. The purpose of this study was to examine the effects of NFE2L1 on the response to oxidative stress in osteoblastic MC3T3-E1 cells. Methods: The murine calvaria-derived MC3T3-E1 cell line was exposed to lipopolysaccharide (LPS) for oxidative stress induc- tion. NFE2L1 effects were evaluated using small interfering RNA (siRNA) for NFE2L1 mRNA. ROS generation and the levels of known antioxidant enzyme genes were assayed. Results: NFE2L1 expression was significantly increased 2.4-fold compared to the control group at 10 μg/mL LPS in MC3T3-E1 cells (P<0.05). LPS increased formation of intracellular ROS in MC3T3-E1 cells. NFE2L1 knockdown led to an additional in- crease of ROS (20%) in the group transfected with NFE2L1 siRNA compared with the control group under LPS stimulation (P<0.05). -

Distinct Isoforms of Nrf1 Diversely Regulate Different Subsets of Its Cognate Target Genes
bioRxiv preprint doi: https://doi.org/10.1101/356071; this version posted June 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Distinct isoforms of Nrf1 diversely regulate different subsets of its cognate target genes Meng Wang, Lu Qiu, Xufang Ru, Yijiang Song, Yiguo Zhang* The Laboratory of Cell Biochemistry and Topogenetic Regulation, College of Bioengineering and Faculty of Sciences, Chongqing University, No. 174 Shazheng Street, Shapingba District, Chongqing 400044, China *Correspondence should be addressed to Yiguo Zhang (email: [email protected] or [email protected]). Abstract The single Nrf1 gene has capability to be differentially transcripted alongside with alternative mRNA-splicing and subsequent translation through different initiation signals so as to yield distinct lengths of polypeptide isoforms. Amongst them, three of the most representatives are Nrf1α, Nrf1β and Nrf1γ, but the putative specific contribution of each isoform to regulating ARE-driven target genes remains unknown. To address this, we have here established three cell lines on the base of the Flp-In™ T-REx™ system, which are allowed for tetracycline-inducibly stable expression of Nrf1α, Nrf1β and Nrf1γ. The RNA-Sequencing results have demonstrated that a vast majority of differentially expressed genes (i.e. 90 DEGs detected) were dominantly up-regulated by Nrf1α and/or Nrf1β following induction by tetracycline. By contrast, other DEGs regulated by Nrf1γ were far less than those regulated by Nrf1α/β (i.e. ~11 of Nrf1α and 7 of Nrf1β). Further transcriptomic analysis revealed that tetracycline-induced expression of Nrf1γ significantly increased the percentage of down-regulated genes in total DEGs. -

Novel Insights Into the Regulation of the Antioxidant Response Element
Novel insights into the regulation of the antioxidant response element mediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by NRF2 Henna-Kaisa Jyrkkänen, Suvi M Kuosmanen, Merja Heinäniemi, Heidi Laitinen, Emilia Kansanen, Eero Mella-Aho, Hanna Leinonen, Seppo Ylä-Herttuala, Anna-Liisa Levonen To cite this version: Henna-Kaisa Jyrkkänen, Suvi M Kuosmanen, Merja Heinäniemi, Heidi Laitinen, Emilia Kansanen, et al.. Novel insights into the regulation of the antioxidant response element mediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by NRF2. Biochemical Journal, Portland Press, 2011, 440 (2), pp.167-174. 10.1042/BJ20110526. hal-00642837 HAL Id: hal-00642837 https://hal.archives-ouvertes.fr/hal-00642837 Submitted on 19 Nov 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 03 Aug 2011 as manuscript BJ20110526 NOVEL INSIGHTS INTO THE REGULATION OF THE ANTIOXIDANT RESPONSE ELEMENT MEDIATED GENE EXPRESSION BY ELECTROPHILES: INDUCTION OF THE TRANSCRIPTIONAL REPRESSOR BACH1 BY NRF2 Henna-Kaisa Jyrkkänen1, Suvi Kuosmanen2, Merja Heinäniemi3, Heidi Laitinen1, Emilia Kansanen1, Eero Mella-Aho1, Hanna Leinonen1, Seppo Ylä-Herttuala1, Anna-Liisa Levonen1* 1Department of Biotechnology and Molecular Medicine, A. -
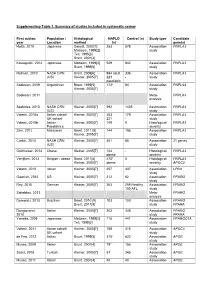
Supplementary Table 1. Summary of Studies Included in Systematic Review
Supplementary Table 1. Summary of studies included in systematic review First author, Population / Histological NAFLD Control (n) Study type Candidate year Location method (n) gene(s) Hotta, 2010 Japanese Sanyal, 2002[1] 253 578 Association PNPLA3 Matteoni, 1999[2] study Teli, 1995[3] Brunt, 2001[4] Kawaguchi, 2012 Japanese Matteoni, 1999[2] 529 942 Association PNPLA3 Brunt, 1999[5] study Rotman, 2010 NASH CRN Brunt, 2009[6] 894 adult 336 Association PNPLA3 (US) Kleiner, 2005[7] 223 - study paediatric Sookoian, 2009 Argentinian Brunt, 1999[5] 172* 94 Association PNPLA3 Kleiner, 2005[7] study Sookoian, 2011 Meta- PNPLA3 analysis Speliotes, 2010 NASH CRN Kleiner, 2005[7] 592 1405 Association PNPLA3 (US) study Valenti, 2010a Italian cohort/ Kleiner, 2005[7] 253 179 Association PNPLA3 UK cohort 321 - study Valenti, 2010b Italian - Kleiner, 2005[7] 149 0 Histological PNPLA3 Paediatrics severity Zain, 2012 Malaysian Brunt, 2011 [8] 144 198 Association PNPLA3 Kleiner, 2005[7] study Corbin, 2013 NASH CRN Kleiner, 2005[7] 361 85 Association 21 genes (US) study Guichelaar, 2013 Obese Kleiner, 2005[7] 144 - Histological PNPLA3 obese severity Verrijken, 2013 Belgian - obese Brunt, 2011[8] 470* 0 Histological PNPLA3 Kleiner, 2005[7] obese severity APOC3 Valenti, 2012 Italian Kleiner, 2005[7] 257 337 Association LPIN1 study Gawrieh, 2012 US Kleiner, 2005[7] 212 62 Association PPARG study Rey, 2010 German Kleiner, 2005[7] 263 259 Healthy Association PPARG 100 AFL study Sahebkar, 2013 Meta- PPARG analysis Domenici, 2013 Brazilian Brunt, 2010 [9] 103 103 -

Genomic Analysis of Allele-Specific Expression in the Mouse Liver
bioRxiv preprint doi: https://doi.org/10.1101/024588; this version posted August 13, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 TITLE: Genomic analysis of allele-specific expression in the mouse liver Ashutosh K. Pandey* and Robert W. Williams* *Department of Genetics, Genomics and Informatics, University of Tennessee Health Science Center, Memphis, TN, 38103 bioRxiv preprint doi: https://doi.org/10.1101/024588; this version posted August 13, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 2 RUNNING TITLE: Allele-specific expression in liver KEYWORDS: BXD, DBA/2J, haplotype-aware alignment, purifying selection, cis eQTL CORRESPONDING AUTHOR: Ashutosh K. Pandey 855 Monroe Avenue, Suite 512 Memphis, TN, 38163 Phone: 901-448-1761 Email addresses: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/024588; this version posted August 13, 2015. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 3 ABSTRACT Genetic differences in gene expression contribute significantly to phenotypic diversity and differences in disease susceptibility. In fact, the great majority of causal variants highlighted by genome-wide association are in non-coding regions that modulate expression. In order to quantify the extent of allelic differences in expression, we analyzed liver transcriptomes of isogenic F1 hybrid mice. Allele-specific expression (ASE) effects are pervasive and are detected in over 50% of assayed genes. -

Snps in Genes Coding for ROS Metabolism and Signalling in Association with Docetaxel Clearance
The Pharmacogenomics Journal (2010) 10, 513–523 & 2010 Macmillan Publishers Limited. All rights reserved 1470-269X/10 www.nature.com/tpj ORIGINAL ARTICLE SNPs in genes coding for ROS metabolism and signalling in association with docetaxel clearance H Edvardsen1,2, PF Brunsvig3, The dose of docetaxel is currently calculated based on body surface area 1,4 5 and does not reflect the pharmacokinetic, metabolic potential or genetic H Solvang , A Tsalenko , background of the patients. The influence of genetic variation on the 6 7 A Andersen , A-C Syvanen , clearance of docetaxel was analysed in a two-stage analysis. In step one, 583 Z Yakhini5, A-L Børresen-Dale1,2, single-nucleotide polymorphisms (SNPs) in 203 genes were genotyped on H Olsen6, S Aamdal3 and samples from 24 patients with locally advanced non-small cell lung cancer. 1,2 We found that many of the genes harbour several SNPs associated with VN Kristensen clearance of docetaxel. Most notably these were four SNPs in EGF, three SNPs 1Department of Genetics, Institute of Cancer in PRDX4 and XPC, and two SNPs in GSTA4, TGFBR2, TNFAIP2, BCL2, DPYD Research, Oslo University Hospital Radiumhospitalet, and EGFR. The multiple SNPs per gene suggested the existence of common Oslo, Norway; 2Institute of Clinical Medicine, haplotypes associated with clearance. These were confirmed with detailed 3 University of Oslo, Oslo, Norway; Cancer Clinic, haplotype analysis. On the basis of analysis of variance (ANOVA), quantitative Oslo University Hospital Radiumhospitalet, Oslo, Norway; 4Institute of -

NRF2-Dependent Gene Expression Promotes Ciliogenesis And
www.nature.com/scientificreports OPEN NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling Received: 8 April 2019 Ana Martin-Hurtado1,2, Raquel Martin-Morales1,2, Natalia Robledinos-Antón1,2,3, Accepted: 11 September 2019 Ruth Blanco1,2,3, Ines Palacios-Blanco1,2, Isabel Lastres-Becker1,2,3, Antonio Cuadrado1,2,3 & Published: xx xx xxxx Francesc R. Garcia-Gonzalo 1,2 The transcription factor NRF2 is a master regulator of cellular antioxidant and detoxifcation responses, but it also regulates other processes such as autophagy and pluripotency. In human embryonic stem cells (hESCs), NRF2 antagonizes neuroectoderm diferentiation, which only occurs after NRF2 is repressed via a Primary Cilia-Autophagy-NRF2 (PAN) axis. However, the functional connections between NRF2 and primary cilia, microtubule-based plasma membrane protrusions that function as cellular antennae, remain poorly understood. For instance, nothing is known about whether NRF2 afects cilia, or whether cilia regulation of NRF2 extends beyond hESCs. Here, we show that NRF2 and primary cilia reciprocally regulate each other. First, we demonstrate that fbroblasts lacking primary cilia have higher NRF2 activity, which is rescued by autophagy-activating mTOR inhibitors, indicating that the PAN axis also operates in diferentiated cells. Furthermore, NRF2 controls cilia formation and function. NRF2-null cells grow fewer and shorter cilia and display impaired Hedgehog signaling, a cilia- dependent pathway. These defects are not due to increased oxidative stress or ciliophagy, but rather to NRF2 promoting expression of multiple ciliogenic and Hedgehog pathway genes. Among these, we focused on GLI2 and GLI3, the transcription factors controlling Hh pathway output. Both their mRNA and protein levels are reduced in NRF2-null cells, consistent with their gene promoters containing consensus ARE sequences predicted to bind NRF2. -

Global Analysis of Somatic Structural Genomic Alterations and Their Impact on Gene Expression in Diverse Human Cancers
Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers Babak Alaei-Mahabadia, Joydeep Bhaduryb, Joakim W. Karlssona, Jonas A. Nilssonb, and Erik Larssona,1 aDepartment of Medical Biochemistry and Cell Biology, Institute of Biomedicine, The Sahlgrenska Academy, University of Gothenburg, SE-405 30 Gothenburg, Sweden; and bDepartment of Surgery, Sahlgrenska Cancer Center, Institute of Clinical Sciences, University of Gothenburg, SE-405 30 Gothenburg, Sweden Edited by Mary-Claire King, University of Washington, Seattle, WA, and approved October 21, 2016 (received for review April 19, 2016) Tumor genomes are mosaics of somatic structural variants (SVs) segments (14). Several factors complicate the analysis, in particular that may contribute to the activation of oncogenes or inactivation mappability issues due to repetitive sequence regions (15). Indeed, of tumor suppressors, for example, by altering gene copy number it has become clear that the results produced by different methods amplitude. However, there are multiple other ways in which SVs are not consistent, and some studies have intersected multiple ap- can modulate transcription, but the general impact of such events proaches to provide a presumed high-confidence set of predictions on tumor transcriptional output has not been systematically de- (16, 17). Adding to the challenges is the difficulty of assessing termined. Here we use whole-genome sequencing data to map SVs performance: True positive sets have thus far been obtained across 600 tumors and 18 cancers, and investigate the relationship through simulated genomic sequences (18), but this will not re- between SVs, copy number alterations (CNAs), and mRNA expression. -

UNIVERSITY of CALIFORNIA Los Angeles Studies on the Role Of
UNIVERSITY OF CALIFORNIA Los Angeles Studies on the role of macrophages in the toxicity induced by diesel exhaust particles, and on cardiovascular effects triggered by electronic cigarettes A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Molecular Toxicology by May Bhetraratana 2018 © Copyright by May Bhetraratana 2018 ABSTRACT OF THE DISSERTATION Studies on the role of macrophages in the toxicity induced by diesel exhaust particles, and on cardiovascular effects triggered by electronic cigarettes by May Bhetraratana Doctor of Philosophy in Molecular Toxicology University of California, Los Angeles, 2018 Professor Jesus A. Araujo, Chair Worldwide, cardiovascular disease is the leading cause of death, with air pollution and smoking being major contributors. Epidemiological studies on air pollution have demonstrated that people living in areas with poorer air quality are at greater risk for hospitalizations, morbidities, and mortality due to cardiovascular and respiratory events. Tobacco cigarette smokers are also at risk for similar events, while users of the increasingly popular electronic cigarette (e-cig) are known to experience symptoms such as coughing and reduced lung function. While much is known about the cardiovascular health effects of air pollution and tobacco cigarette smoking, though, there remain important knowledge gaps – (1) what are the molecular mechanisms linking air pollution with disease that extends beyond the lungs, and (2) are e-cig users still at risk for developing cardiovascular disease like tobacco smokers? ii Our research with a model air pollutant, disease exhaust particles (DEP), therefore focused on the role of a particular cell type, the macrophage, in the mechanism behind the toxicity of air pollution. -

Early Loss of Mitochondrial Complex I and Rewiring of Glutathione
Early loss of mitochondrial complex I and rewiring of PNAS PLUS glutathione metabolism in renal oncocytoma Raj K. Gopala,b,c,d, Sarah E. Calvoa,b,c, Angela R. Shihe, Frances L. Chavesf, Declan McGuonee, Eran Micka,b,c, Kerry A. Piercec, Yang Lia,g, Andrea Garofaloc,h, Eliezer M. Van Allenc,h, Clary B. Clishc, Esther Olivae, and Vamsi K. Moothaa,b,c,1 aHoward Hughes Medical Institute and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114; bDepartment of Systems Biology, Harvard Medical School, Boston, MA 02115; cBroad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Hematology/Oncology, Massachusetts General Hospital, Boston, MA 02114; eDepartment of Pathology, Massachusetts General Hospital, Boston, MA 02114; fMolecular Pathology Unit, Massachusetts General Hospital, Boston, MA 02114; gDepartment of Statistics, Harvard University, Cambridge, MA 02138; and hDepartment of Medical Oncology, Dana–Farber Cancer Institute, Boston, MA 02215 Contributed by Vamsi K. Mootha, April 17, 2018 (sent for review July 6, 2017; reviewed by Ralph J. DeBerardinis and Robert A. Weinberg) Renal oncocytomas are benign tumors characterized by a marked electron transport chain (10, 11). Sequencing of nuclear DNA in accumulation of mitochondria. We report a combined exome, RO has revealed a low somatic mutation rate without recurrent transcriptome, and metabolome analysis of these tumors. Joint nuclear gene mutations, although recurrent chromosome 1 loss analysis of the nuclear and mitochondrial (mtDNA) genomes and cyclin D1 rearrangement have been reported (12, 13). reveals loss-of-function mtDNA mutations occurring at high vari- In this study, we report the results of profiling DNA, RNA, ant allele fractions, consistent with positive selection, in genes and metabolites in RO to investigate the molecular and bio- encoding complex I as the most frequent genetic events.