Inhibition of Thioredoxin/Thioredoxin Reductase Induces Synthetic Lethality in Lung
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

Viewed BMC Genomics 2001, 2:10
BMC Genomics BioMed Central BMC Genomics :10 Research2 2001, article Genomic organisation and alternative splicing of mouse and human thioredoxin reductase 1 genes Simone A Osborne and Kathryn F Tonissen* Address: School of Biomolecular and Biomedical Science, Griffith University, Nathan, Queensland 4111, Australia E-mail: Simone A Osborne - [email protected]; Kathryn F Tonissen* - [email protected] *Corresponding author Published: 22 November 2001 Received: 28 August 2001 Accepted: 22 November 2001 BMC Genomics 2001, 2:10 This article is available from: http://www.biomedcentral.com/1471-2164/2/10 © 2001 Osborne and Tonissen; licensee BioMed Central Ltd. Verbatim copying and redistribution of this article are permitted in any medium for any non-commercial purpose, provided this notice is preserved along with the article's original URL. For commercial use, contact [email protected] Abstract Background: Thioredoxin reductase (TR) is a redox active protein involved in many cellular processes as part of the thioredoxin system. Presently there are three recognised forms of mammalian thioredoxin reductase designated as TR1, TR3 and TGR, that represent the cytosolic, mitochondrial and novel forms respectively. In this study we elucidated the genomic organisation of the mouse (Txnrd1) and human thioredoxin reductase 1 genes (TXNRD1) through library screening, restriction mapping and database mining. Results: The human TXNRD1 gene spans 100 kb of genomic DNA organised into 16 exons and the mouse Txnrd1 gene has a similar exon/intron arrangement. We also analysed the alternative splicing patterns displayed by the mouse and human thioredoxin reductase 1 genes and mapped the different mRNA isoforms with respect to genomic organisation. -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells
Original Endocrinol Metab 2016;31:336-342 http://dx.doi.org/10.3803/EnM.2016.31.2.336 Article pISSN 2093-596X · eISSN 2093-5978 The Role of Nuclear Factor-E2-Related Factor 1 in the Oxidative Stress Response in MC3T3-E1 Osteoblastic Cells So Young Park1, Sung Hoon Kim1, Hyun Koo Yoon1, Chang Hoon Yim1, Sung-Kil Lim2 1Department of Internal Medicine, Cheil General Hospital & Women’s Healthcare Center, Dankook University College of Medicine, Seoul; 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea Background: Reactive oxygen species (ROS) and antioxidants are associated with maintenance of cellular function and metabo- lism. Nuclear factor-E2-related factor 1 (NFE2L1, Nrf1) is known to regulate the expression of a number of genes involved in oxidative stress and inflammation. The purpose of this study was to examine the effects of NFE2L1 on the response to oxidative stress in osteoblastic MC3T3-E1 cells. Methods: The murine calvaria-derived MC3T3-E1 cell line was exposed to lipopolysaccharide (LPS) for oxidative stress induc- tion. NFE2L1 effects were evaluated using small interfering RNA (siRNA) for NFE2L1 mRNA. ROS generation and the levels of known antioxidant enzyme genes were assayed. Results: NFE2L1 expression was significantly increased 2.4-fold compared to the control group at 10 μg/mL LPS in MC3T3-E1 cells (P<0.05). LPS increased formation of intracellular ROS in MC3T3-E1 cells. NFE2L1 knockdown led to an additional in- crease of ROS (20%) in the group transfected with NFE2L1 siRNA compared with the control group under LPS stimulation (P<0.05). -

University of Cincinnati
UNIVERSITY OF CINCINNATI _____________ , 20 _____ I,______________________________________________, hereby submit this as part of the requirements for the degree of: ________________________________________________ in: ________________________________________________ It is entitled: ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ Approved by: ________________________ ________________________ ________________________ ________________________ ________________________ Role of Glutamate-Cysteine Ligase in Maintaining Glutathione Homeostasis and Protecting against Oxidative Stress A dissertation submitted to the Division of Research and Advanced Studies University of Cincinnati in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (Ph.D.) in the Department of Environmental Health of the College of Medicine 2003 by Yi Yang M.D., Sun Yat-sen University of Medical Sciences, 1995 M.S., Sun Yat-sen University of Medical Sciences, 1997 Committee Chair: Daniel W. Nebert, M.D. Professor Department of Environmental Health University of Cincinnati Abstract Glutamate-cysteine ligase (GCL) is the rate-limiting enzyme catalyzing the first step of glutathione (GSH) biosynthesis. In higher eukaryotes, this enzyme is a heterodimer comprising a catalytic subunit (GCLC) and a modifier subunit (GCLM); the latter changes the catalytic characteristics of the holoenzyme. In the first part -

Distinct Isoforms of Nrf1 Diversely Regulate Different Subsets of Its Cognate Target Genes
bioRxiv preprint doi: https://doi.org/10.1101/356071; this version posted June 28, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Distinct isoforms of Nrf1 diversely regulate different subsets of its cognate target genes Meng Wang, Lu Qiu, Xufang Ru, Yijiang Song, Yiguo Zhang* The Laboratory of Cell Biochemistry and Topogenetic Regulation, College of Bioengineering and Faculty of Sciences, Chongqing University, No. 174 Shazheng Street, Shapingba District, Chongqing 400044, China *Correspondence should be addressed to Yiguo Zhang (email: [email protected] or [email protected]). Abstract The single Nrf1 gene has capability to be differentially transcripted alongside with alternative mRNA-splicing and subsequent translation through different initiation signals so as to yield distinct lengths of polypeptide isoforms. Amongst them, three of the most representatives are Nrf1α, Nrf1β and Nrf1γ, but the putative specific contribution of each isoform to regulating ARE-driven target genes remains unknown. To address this, we have here established three cell lines on the base of the Flp-In™ T-REx™ system, which are allowed for tetracycline-inducibly stable expression of Nrf1α, Nrf1β and Nrf1γ. The RNA-Sequencing results have demonstrated that a vast majority of differentially expressed genes (i.e. 90 DEGs detected) were dominantly up-regulated by Nrf1α and/or Nrf1β following induction by tetracycline. By contrast, other DEGs regulated by Nrf1γ were far less than those regulated by Nrf1α/β (i.e. ~11 of Nrf1α and 7 of Nrf1β). Further transcriptomic analysis revealed that tetracycline-induced expression of Nrf1γ significantly increased the percentage of down-regulated genes in total DEGs. -
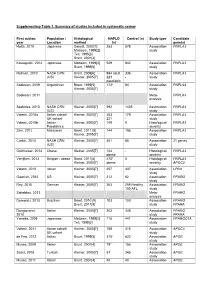
Supplementary Table 1. Summary of Studies Included in Systematic Review
Supplementary Table 1. Summary of studies included in systematic review First author, Population / Histological NAFLD Control (n) Study type Candidate year Location method (n) gene(s) Hotta, 2010 Japanese Sanyal, 2002[1] 253 578 Association PNPLA3 Matteoni, 1999[2] study Teli, 1995[3] Brunt, 2001[4] Kawaguchi, 2012 Japanese Matteoni, 1999[2] 529 942 Association PNPLA3 Brunt, 1999[5] study Rotman, 2010 NASH CRN Brunt, 2009[6] 894 adult 336 Association PNPLA3 (US) Kleiner, 2005[7] 223 - study paediatric Sookoian, 2009 Argentinian Brunt, 1999[5] 172* 94 Association PNPLA3 Kleiner, 2005[7] study Sookoian, 2011 Meta- PNPLA3 analysis Speliotes, 2010 NASH CRN Kleiner, 2005[7] 592 1405 Association PNPLA3 (US) study Valenti, 2010a Italian cohort/ Kleiner, 2005[7] 253 179 Association PNPLA3 UK cohort 321 - study Valenti, 2010b Italian - Kleiner, 2005[7] 149 0 Histological PNPLA3 Paediatrics severity Zain, 2012 Malaysian Brunt, 2011 [8] 144 198 Association PNPLA3 Kleiner, 2005[7] study Corbin, 2013 NASH CRN Kleiner, 2005[7] 361 85 Association 21 genes (US) study Guichelaar, 2013 Obese Kleiner, 2005[7] 144 - Histological PNPLA3 obese severity Verrijken, 2013 Belgian - obese Brunt, 2011[8] 470* 0 Histological PNPLA3 Kleiner, 2005[7] obese severity APOC3 Valenti, 2012 Italian Kleiner, 2005[7] 257 337 Association LPIN1 study Gawrieh, 2012 US Kleiner, 2005[7] 212 62 Association PPARG study Rey, 2010 German Kleiner, 2005[7] 263 259 Healthy Association PPARG 100 AFL study Sahebkar, 2013 Meta- PPARG analysis Domenici, 2013 Brazilian Brunt, 2010 [9] 103 103 -

Snps in Genes Coding for ROS Metabolism and Signalling in Association with Docetaxel Clearance
The Pharmacogenomics Journal (2010) 10, 513–523 & 2010 Macmillan Publishers Limited. All rights reserved 1470-269X/10 www.nature.com/tpj ORIGINAL ARTICLE SNPs in genes coding for ROS metabolism and signalling in association with docetaxel clearance H Edvardsen1,2, PF Brunsvig3, The dose of docetaxel is currently calculated based on body surface area 1,4 5 and does not reflect the pharmacokinetic, metabolic potential or genetic H Solvang , A Tsalenko , background of the patients. The influence of genetic variation on the 6 7 A Andersen , A-C Syvanen , clearance of docetaxel was analysed in a two-stage analysis. In step one, 583 Z Yakhini5, A-L Børresen-Dale1,2, single-nucleotide polymorphisms (SNPs) in 203 genes were genotyped on H Olsen6, S Aamdal3 and samples from 24 patients with locally advanced non-small cell lung cancer. 1,2 We found that many of the genes harbour several SNPs associated with VN Kristensen clearance of docetaxel. Most notably these were four SNPs in EGF, three SNPs 1Department of Genetics, Institute of Cancer in PRDX4 and XPC, and two SNPs in GSTA4, TGFBR2, TNFAIP2, BCL2, DPYD Research, Oslo University Hospital Radiumhospitalet, and EGFR. The multiple SNPs per gene suggested the existence of common Oslo, Norway; 2Institute of Clinical Medicine, haplotypes associated with clearance. These were confirmed with detailed 3 University of Oslo, Oslo, Norway; Cancer Clinic, haplotype analysis. On the basis of analysis of variance (ANOVA), quantitative Oslo University Hospital Radiumhospitalet, Oslo, Norway; 4Institute of -

NRF2-Dependent Gene Expression Promotes Ciliogenesis And
www.nature.com/scientificreports OPEN NRF2-dependent gene expression promotes ciliogenesis and Hedgehog signaling Received: 8 April 2019 Ana Martin-Hurtado1,2, Raquel Martin-Morales1,2, Natalia Robledinos-Antón1,2,3, Accepted: 11 September 2019 Ruth Blanco1,2,3, Ines Palacios-Blanco1,2, Isabel Lastres-Becker1,2,3, Antonio Cuadrado1,2,3 & Published: xx xx xxxx Francesc R. Garcia-Gonzalo 1,2 The transcription factor NRF2 is a master regulator of cellular antioxidant and detoxifcation responses, but it also regulates other processes such as autophagy and pluripotency. In human embryonic stem cells (hESCs), NRF2 antagonizes neuroectoderm diferentiation, which only occurs after NRF2 is repressed via a Primary Cilia-Autophagy-NRF2 (PAN) axis. However, the functional connections between NRF2 and primary cilia, microtubule-based plasma membrane protrusions that function as cellular antennae, remain poorly understood. For instance, nothing is known about whether NRF2 afects cilia, or whether cilia regulation of NRF2 extends beyond hESCs. Here, we show that NRF2 and primary cilia reciprocally regulate each other. First, we demonstrate that fbroblasts lacking primary cilia have higher NRF2 activity, which is rescued by autophagy-activating mTOR inhibitors, indicating that the PAN axis also operates in diferentiated cells. Furthermore, NRF2 controls cilia formation and function. NRF2-null cells grow fewer and shorter cilia and display impaired Hedgehog signaling, a cilia- dependent pathway. These defects are not due to increased oxidative stress or ciliophagy, but rather to NRF2 promoting expression of multiple ciliogenic and Hedgehog pathway genes. Among these, we focused on GLI2 and GLI3, the transcription factors controlling Hh pathway output. Both their mRNA and protein levels are reduced in NRF2-null cells, consistent with their gene promoters containing consensus ARE sequences predicted to bind NRF2. -

Global Analysis of Somatic Structural Genomic Alterations and Their Impact on Gene Expression in Diverse Human Cancers
Global analysis of somatic structural genomic alterations and their impact on gene expression in diverse human cancers Babak Alaei-Mahabadia, Joydeep Bhaduryb, Joakim W. Karlssona, Jonas A. Nilssonb, and Erik Larssona,1 aDepartment of Medical Biochemistry and Cell Biology, Institute of Biomedicine, The Sahlgrenska Academy, University of Gothenburg, SE-405 30 Gothenburg, Sweden; and bDepartment of Surgery, Sahlgrenska Cancer Center, Institute of Clinical Sciences, University of Gothenburg, SE-405 30 Gothenburg, Sweden Edited by Mary-Claire King, University of Washington, Seattle, WA, and approved October 21, 2016 (received for review April 19, 2016) Tumor genomes are mosaics of somatic structural variants (SVs) segments (14). Several factors complicate the analysis, in particular that may contribute to the activation of oncogenes or inactivation mappability issues due to repetitive sequence regions (15). Indeed, of tumor suppressors, for example, by altering gene copy number it has become clear that the results produced by different methods amplitude. However, there are multiple other ways in which SVs are not consistent, and some studies have intersected multiple ap- can modulate transcription, but the general impact of such events proaches to provide a presumed high-confidence set of predictions on tumor transcriptional output has not been systematically de- (16, 17). Adding to the challenges is the difficulty of assessing termined. Here we use whole-genome sequencing data to map SVs performance: True positive sets have thus far been obtained across 600 tumors and 18 cancers, and investigate the relationship through simulated genomic sequences (18), but this will not re- between SVs, copy number alterations (CNAs), and mRNA expression. -

Upregulation of Thioredoxin Reductase 1 in Human Oral Squamous Cell Carcinoma
637-644.qxd 19/1/2011 09:58 Ì ™ÂÏ›‰·637 ONCOLOGY REPORTS 25: 637-644, 2011 637 Upregulation of thioredoxin reductase 1 in human oral squamous cell carcinoma SHUNICHIRO IWASAWA1, YUKIO YAMANO2, YUICHI TAKIGUCHI1, HIDEKI TANZAWA2,3, KOICHIRO TATSUMI1 and KATSUHIRO UZAWA2,3 Departments of 1Respirology, and 2Clinical Molecular Biology, Graduate School of Medicine, Chiba University; 3Division of Dentistry and Oral-Maxillofacial Surgery, Chiba University Hospital, 1-8-1 Inohana, Chuo-ku, Chiba 260-8670, Japan Received September 3, 2010; Accepted October 29, 2010 DOI: 10.3892/or.2010.1131 Abstract. Thioredoxin reductase 1 (TrxR1) catalyzes the nico- advances in surgical and radiation therapies in recent decades, tinamide adenine dinucleotide phosphate-dependent reduction patients diagnosed with stages I and II have a relatively of oxidized thioredoxin (Trx). Trx, which is over-expressed in good prognosis. However, patients with stages III and IV, many human tumors, is a selenocysteine-containing protein which account for more than two-thirds of cases, have a high associated with cell proliferation and apoptosis inhibition. recurrence rate at regional and distal sites of metastasis This selenium-containing redox system regulates the activity (6,7). The resulting survival rates of all patients with OSCC of various enzymes and counteracts oxidative stress in cells remain poor. To improve the prognosis, strategies have such as hypoxia and cytotoxic agents. Consequently, TrxR1 been developed to integrate systemic chemotherapy into the could play an important role in tumor progression and resis- perioperative period. Moreover, molecular targeted therapy tance to chemotherapy due to its anti-apoptotic functions. To recently has been extensively investigated as a single modality characterize cancer-related gene expression changes in oral and in combination with cytotoxic treatments (8). -

British Journal of Nutrition (2014), 112, 295–308 Doi:10.1017/S0007114514000841 Q the Author 2014
Downloaded from British Journal of Nutrition (2014), 112, 295–308 doi:10.1017/S0007114514000841 q The Author 2014 https://www.cambridge.org/core Effect of dietary a-lipoic acid on the mRNA expression of genes involved in drug metabolism and antioxidation system in rat liver Takashi Ide*† . IP address: Laboratory of Nutritional Function, National Food Research Institute, 2-1-12 Kannondai, Tsukuba 305-8642, Japan (Submitted 30 July 2013 – Final revision received 11 March 2014 – Accepted 20 March 2014 – First published online 1 May 2014) 170.106.202.226 Abstract In the present study, the mRNA levels of hepatic proteins involved in the drug metabolism of rats fed a-lipoic acid were evaluated by DNA , on microarray and real-time PCR analyses. Experimental diets containing 0, 0·1, 0·25 and 0·5 % (w/w) a-lipoic acid were fed to four groups of 28 Sep 2021 at 21:50:59 rats consisting of seven animals each for 21 d. DNA microarray analysis revealed that the diet containing 0·5 % a-lipoic acid significantly (P,0·05) increased the mRNA levels of various phase I drug-metabolising enzymes up to 15-fold and phase II enzymes up to 52-fold in an isoenzyme-specific manner. a-Lipoic acid also up-regulated the mRNA levels of some members of the ATP-binding cassette transpor- ter superfamily, presumed to be involved in the exportation of xenobiotics, up to 6·6-fold. In addition, we observed that a-lipoic acid increased the mRNA levels of many proteins involved in antioxidation, such as members of the thiol redox system (up to 5·5-fold), metal- lothioneins (up to 12-fold) and haeme oxygenase 1 (1·5-fold).