NRF2-Dependent Gene Expression Promotes Ciliogenesis And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

The Title of the Dissertation
UNIVERSITY OF CALIFORNIA SAN DIEGO Novel network-based integrated analyses of multi-omics data reveal new insights into CD8+ T cell differentiation and mouse embryogenesis A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Bioinformatics and Systems Biology by Kai Zhang Committee in charge: Professor Wei Wang, Chair Professor Pavel Arkadjevich Pevzner, Co-Chair Professor Vineet Bafna Professor Cornelis Murre Professor Bing Ren 2018 Copyright Kai Zhang, 2018 All rights reserved. The dissertation of Kai Zhang is approved, and it is accept- able in quality and form for publication on microfilm and electronically: Co-Chair Chair University of California San Diego 2018 iii EPIGRAPH The only true wisdom is in knowing you know nothing. —Socrates iv TABLE OF CONTENTS Signature Page ....................................... iii Epigraph ........................................... iv Table of Contents ...................................... v List of Figures ........................................ viii List of Tables ........................................ ix Acknowledgements ..................................... x Vita ............................................. xi Abstract of the Dissertation ................................. xii Chapter 1 General introduction ............................ 1 1.1 The applications of graph theory in bioinformatics ......... 1 1.2 Leveraging graphs to conduct integrated analyses .......... 4 1.3 References .............................. 6 Chapter 2 Systematic -

The Autophagy Receptor SQSTM1/P62 Mediates Anti-Inflammatory Actions of the Selective NR3C1/ Glucocorticoid Receptor Modulator Compound a (Cpda) in Macrophages
Autophagy ISSN: 1554-8627 (Print) 1554-8635 (Online) Journal homepage: http://www.tandfonline.com/loi/kaup20 The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/ glucocorticoid receptor modulator compound A (CpdA) in macrophages Viacheslav Mylka, Julie Deckers, Dariusz Ratman, Lode De Cauwer, Jonathan Thommis, Riet De Rycke, Francis Impens, Claude Libert, Jan Tavernier, Wim Vanden Berghe, Kris Gevaert & Karolien De Bosscher To cite this article: Viacheslav Mylka, Julie Deckers, Dariusz Ratman, Lode De Cauwer, Jonathan Thommis, Riet De Rycke, Francis Impens, Claude Libert, Jan Tavernier, Wim Vanden Berghe, Kris Gevaert & Karolien De Bosscher (2018) The autophagy receptor SQSTM1/p62 mediates anti- inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator compound A (CpdA) in macrophages, Autophagy, 14:12, 2049-2064, DOI: 10.1080/15548627.2018.1495681 To link to this article: https://doi.org/10.1080/15548627.2018.1495681 © 2018 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. Published online: 14 Sep 2018. Submit your article to this journal Article views: 907 View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=kaup20 AUTOPHAGY 2018, VOL. 14, NO. 12, 2049–2064 https://doi.org/10.1080/15548627.2018.1495681 RESEARCH PAPER - BASIC SCIENCE The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator -

Regulation of Adult Neurogenesis in Mammalian Brain
International Journal of Molecular Sciences Review Regulation of Adult Neurogenesis in Mammalian Brain 1,2, 3, 3,4 Maria Victoria Niklison-Chirou y, Massimiliano Agostini y, Ivano Amelio and Gerry Melino 3,* 1 Centre for Therapeutic Innovation (CTI-Bath), Department of Pharmacy & Pharmacology, University of Bath, Bath BA2 7AY, UK; [email protected] 2 Blizard Institute of Cell and Molecular Science, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London E1 2AT, UK 3 Department of Experimental Medicine, TOR, University of Rome “Tor Vergata”, 00133 Rome, Italy; [email protected] (M.A.); [email protected] (I.A.) 4 School of Life Sciences, University of Nottingham, Nottingham NG7 2HU, UK * Correspondence: [email protected] These authors contributed equally to this work. y Received: 18 May 2020; Accepted: 7 July 2020; Published: 9 July 2020 Abstract: Adult neurogenesis is a multistage process by which neurons are generated and integrated into existing neuronal circuits. In the adult brain, neurogenesis is mainly localized in two specialized niches, the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) adjacent to the lateral ventricles. Neurogenesis plays a fundamental role in postnatal brain, where it is required for neuronal plasticity. Moreover, perturbation of adult neurogenesis contributes to several human diseases, including cognitive impairment and neurodegenerative diseases. The interplay between extrinsic and intrinsic factors is fundamental in regulating neurogenesis. Over the past decades, several studies on intrinsic pathways, including transcription factors, have highlighted their fundamental role in regulating every stage of neurogenesis. However, it is likely that transcriptional regulation is part of a more sophisticated regulatory network, which includes epigenetic modifications, non-coding RNAs and metabolic pathways. -

Expression Profiling of KLF4
Expression Profiling of KLF4 AJCR0000006 Supplemental Data Figure S1. Snapshot of enriched gene sets identified by GSEA in Klf4-null MEFs. Figure S2. Snapshot of enriched gene sets identified by GSEA in wild type MEFs. 98 Am J Cancer Res 2011;1(1):85-97 Table S1: Functional Annotation Clustering of Genes Up-Regulated in Klf4 -Null MEFs ILLUMINA_ID Gene Symbol Gene Name (Description) P -value Fold-Change Cell Cycle 8.00E-03 ILMN_1217331 Mcm6 MINICHROMOSOME MAINTENANCE DEFICIENT 6 40.36 ILMN_2723931 E2f6 E2F TRANSCRIPTION FACTOR 6 26.8 ILMN_2724570 Mapk12 MITOGEN-ACTIVATED PROTEIN KINASE 12 22.19 ILMN_1218470 Cdk2 CYCLIN-DEPENDENT KINASE 2 9.32 ILMN_1234909 Tipin TIMELESS INTERACTING PROTEIN 5.3 ILMN_1212692 Mapk13 SAPK/ERK/KINASE 4 4.96 ILMN_2666690 Cul7 CULLIN 7 2.23 ILMN_2681776 Mapk6 MITOGEN ACTIVATED PROTEIN KINASE 4 2.11 ILMN_2652909 Ddit3 DNA-DAMAGE INDUCIBLE TRANSCRIPT 3 2.07 ILMN_2742152 Gadd45a GROWTH ARREST AND DNA-DAMAGE-INDUCIBLE 45 ALPHA 1.92 ILMN_1212787 Pttg1 PITUITARY TUMOR-TRANSFORMING 1 1.8 ILMN_1216721 Cdk5 CYCLIN-DEPENDENT KINASE 5 1.78 ILMN_1227009 Gas2l1 GROWTH ARREST-SPECIFIC 2 LIKE 1 1.74 ILMN_2663009 Rassf5 RAS ASSOCIATION (RALGDS/AF-6) DOMAIN FAMILY 5 1.64 ILMN_1220454 Anapc13 ANAPHASE PROMOTING COMPLEX SUBUNIT 13 1.61 ILMN_1216213 Incenp INNER CENTROMERE PROTEIN 1.56 ILMN_1256301 Rcc2 REGULATOR OF CHROMOSOME CONDENSATION 2 1.53 Extracellular Matrix 5.80E-06 ILMN_2735184 Col18a1 PROCOLLAGEN, TYPE XVIII, ALPHA 1 51.5 ILMN_1223997 Crtap CARTILAGE ASSOCIATED PROTEIN 32.74 ILMN_2753809 Mmp3 MATRIX METALLOPEPTIDASE -

Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors
cancers Review Roles of NRF3 in the Hallmarks of Cancer: Proteasomal Inactivation of Tumor Suppressors Akira Kobayashi 1,2 1 Laboratory for Genetic Code, Graduate School of Life and Medical Sciences, Doshisha University, Kyotanabe, Kyoto 610-0394, Japan; [email protected]; Tel.: +81-774-65-6273 2 Department of Medical Life Systems, Faculty of Life and Medical Sciences, Doshisha University, Kyotanabe, Kyoto 610-0394, Japan Received: 1 September 2020; Accepted: 17 September 2020; Published: 20 September 2020 Simple Summary: This review summarizes recent advances in our understanding of the physiological roles of the NFE2-related factor 2 (NRF2)-related transcription factor NRF3 in cancer. NRF3 confers cells with six so-called “hallmarks of cancer” through upregulating gene expression of specific target genes, leading to tumorigenesis and cancer malignancy. These driver gene-like functions of NRF3 in cancer are distinct from those of NRF2. Abstract: The physiological roles of the NRF2-related transcription factor NRF3 (NFE2L3) have remained unknown for decades. The remarkable development of human cancer genome databases has led to strong suggestions that NRF3 has functional significance in cancer; specifically,high NRF3 mRNA levels are induced in many cancer types, such as colorectal cancer and pancreatic adenocarcinoma, and are associated with poor prognosis. On the basis of this information, the involvement of NRF3 in tumorigenesis and cancer malignancy has been recently proposed. NRF3 confers cancer cells with selective growth advantages by enhancing 20S proteasome assembly through induction of the chaperone gene proteasome maturation protein (POMP) and consequently promoting degradation of the tumor suppressors p53 and retinoblastoma (Rb) in a ubiquitin-independent manner. -

Genome-Scale Identification of Transcription Factors That Mediate An
ARTICLE DOI: 10.1038/s41467-018-04406-2 OPEN Genome-scale identification of transcription factors that mediate an inflammatory network during breast cellular transformation Zhe Ji 1,2,4, Lizhi He1, Asaf Rotem1,2,5, Andreas Janzer1,6, Christine S. Cheng2,7, Aviv Regev2,3 & Kevin Struhl 1 Transient activation of Src oncoprotein in non-transformed, breast epithelial cells can initiate an epigenetic switch to the stably transformed state via a positive feedback loop that involves 1234567890():,; the inflammatory transcription factors STAT3 and NF-κB. Here, we develop an experimental and computational pipeline that includes 1) a Bayesian network model (AccessTF) that accurately predicts protein-bound DNA sequence motifs based on chromatin accessibility, and 2) a scoring system (TFScore) that rank-orders transcription factors as candidates for being important for a biological process. Genetic experiments validate TFScore and suggest that more than 40 transcription factors contribute to the oncogenic state in this model. Interestingly, individual depletion of several of these factors results in similar transcriptional profiles, indicating that a complex and interconnected transcriptional network promotes a stable oncogenic state. The combined experimental and computational pipeline represents a general approach to comprehensively identify transcriptional regulators important for a biological process. 1 Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115, USA. 2 Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA. 3 Department of Biology, Howard Hughes Medical Institute and David H. Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, MA 20140, USA. 4Present address: Department of Pharmacology and Biomedical Engineering, Northwestern University, Evanston 60611 IL, USA. -

NFE2L1 Antibody Cat
NFE2L1 Antibody Cat. No.: 31-335 NFE2L1 Antibody Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse, Rat Antibody produced in rabbits immunized with a synthetic peptide corresponding a region IMMUNOGEN: of human NFE2L1. TESTED APPLICATIONS: ELISA, WB NFE2L1 antibody can be used for detection of NFE2L1 by ELISA at 1:7862500. NFE2L1 APPLICATIONS: antibody can be used for detection of NFE2L1 by western blot at 1 μg/mL, and HRP conjugated secondary antibody should be diluted 1:50,000 - 100,000. POSITIVE CONTROL: 1) Cat. No. 1205 - Jurkat Cell Lysate PREDICTED MOLECULAR 85 kDa WEIGHT: Properties PURIFICATION: Antibody is purified by peptide affinity chromatography method. CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid September 27, 2021 1 https://www.prosci-inc.com/nfe2l1-antibody-31-335.html Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% BUFFER: sucrose. CONCENTRATION: batch dependent For short periods of storage (days) store at 4˚C. For longer periods of storage, store STORAGE CONDITIONS: NFE2L1 antibody at -20˚C. As with any antibody avoid repeat freeze-thaw cycles. Additional Info OFFICIAL SYMBOL: NFE2L1 ALTERNATE NAMES: NFE2L1, FLJ00380, LCR-F1, NRF1, TCF11 ACCESSION NO.: NP_003195 PROTEIN GI NO.: 4505379 GENE ID: 4779 USER NOTE: Optimal dilutions for each application to be determined by the researcher. Background and References NFE2L1 activates erythroid-specific, globin gene expression. This gene encodes a protein that is involved in globin gene expression in erythrocytes. Confusion has occurred in bibliographic databases due to the shared symbol of NRF1 for this gene, NFE2L1, and for BACKGROUND: 'nuclear respiratory factor 1' which has an official symbol of NRF1. -

Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition
Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 RESEARCH ARTICLE Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition Alexandre Puissant1,3, Stacey M. Frumm1,3, Gabriela Alexe1,3,5,6, Christopher F. Bassil1,3, Jun Qi2, Yvan H. Chanthery8, Erin A. Nekritz8, Rhamy Zeid2, William Clay Gustafson8, Patricia Greninger7, Matthew J. Garnett10, Ultan McDermott10, Cyril H. Benes7, Andrew L. Kung1,3, William A. Weiss8,9, James E. Bradner2,4, and Kimberly Stegmaier1,3,6 Downloaded from cancerdiscovery.aacrjournals.org on October 2, 2021. © 2013 American Association for Cancer Research. 15-CD-12-0418_p308-323.indd 1 22/02/13 12:15 AM Published OnlineFirst February 21, 2013; DOI: 10.1158/2159-8290.CD-12-0418 A BSTRACT Bromodomain inhibition comprises a promising therapeutic strategy in cancer, particularly for hematologic malignancies. To date, however, genomic biomarkers to direct clinical translation have been lacking. We conducted a cell-based screen of genetically defined cancer cell lines using a prototypical inhibitor of BET bromodomains. Integration of genetic features with chemosensitivity data revealed a robust correlation between MYCN amplification and sensitivity to bromodomain inhibition. We characterized the mechanistic and translational significance of this finding in neuroblastoma, a childhood cancer with frequent amplification of MYCN. Genome-wide expression analysis showed downregulation of the MYCN transcriptional program accompanied by suppression of MYCN transcription. Functionally, bromodomain-mediated inhibition of MYCN impaired growth and induced apoptosis in neuroblastoma. BRD4 knockdown phenocopied these effects, establishing BET bromodomains as transcriptional regulators of MYCN. BET inhibition conferred a significant survival advantage in 3 in vivo neuroblastoma models, providing a compelling rationale for developing BET bro- modomain inhibitors in patients with neuroblastoma. -

The Suppressive Effects of 1,25-Dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Masters Theses Graduate School 8-2014 The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration Carolyn Jeanne Ricciardi University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_gradthes Part of the Molecular, Genetic, and Biochemical Nutrition Commons Recommended Citation Ricciardi, Carolyn Jeanne, "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration. " Master's Thesis, University of Tennessee, 2014. https://trace.tennessee.edu/utk_gradthes/2876 This Thesis is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Masters Theses by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a thesis written by Carolyn Jeanne Ricciardi entitled "The Suppressive Effects of 1,25-dihydroxyvitamin D3 and Vitamin D Receptor on Brown Adipocyte Differentiation and Mitochondrial Respiration." I have examined the final electronic copy of this thesis for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Master of Science, with a major in Nutrition. -

High-Throughput Bioinformatics Approaches to Understand Gene Expression Regulation in Head and Neck Tumors
High-throughput bioinformatics approaches to understand gene expression regulation in head and neck tumors by Yanxiao Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Bioinformatics) in The University of Michigan 2016 Doctoral Committee: Associate Professor Maureen A. Sartor, Chair Professor Thomas E. Carey Assistant Professor Hui Jiang Professor Ronald J. Koenig Associate Professor Laura M. Rozek Professor Kerby A. Shedden c Yanxiao Zhang 2016 All Rights Reserved I dedicate this thesis to my family. For their unfailing love, understanding and support. ii ACKNOWLEDGEMENTS I would like to express my gratitude to Dr. Maureen Sartor for her guidance in my research and career development. She is a great mentor. She patiently taught me when I started new in this field, granted me freedom to explore and helped me out when I got lost. Her dedication to work, enthusiasm in teaching, mentoring and communicating science have inspired me to feel the excite- ment of research beyond novel scientific discoveries. I’m also grateful to have an interdisciplinary committee. Their feedback on my research progress and presentation skills is very valuable. In particular, I would like to thank Dr. Thomas Carey and Dr. Laura Rozek for insightful discussions on the biology of head and neck cancers and human papillomavirus, Dr. Ronald Koenig for expert knowledge on thyroid cancers, Dr. Hui Jiang and Dr. Kerby Shedden for feedback on the statistics part of my thesis. I would like to thank all the past and current members of Sartor lab for making the lab such a lovely place to stay and work in. -
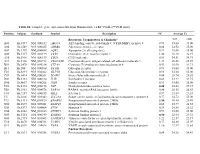
Table S1. Complete Gene Expression Data from Human Diabetes RT² Profiler™ PCR Array Receptors, Transporters & Channels* A
Table S1. Complete gene expression data from Human Diabetes RT² Profiler™ PCR Array Position Unigene GenBank Symbol Description FC Average Ct Receptors, Transporters & Channels* NGT GDM A01 Hs,5447 NM_000352 ABCC8 ATP-binding cassette, sub-family C (CFTR/MRP), member 8 0.93 35.00 35.00 A04 0Hs,2549 NM_000025 ADRB3 Adrenergic, beta-3-, receptor 0.88 34.92 35.00 A07 Hs,1307 NM_000486 AQP2 Aquaporin 2 (collecting duct) 0.93 35.00 35.00 A09 30Hs,5117 NM_001123 CCR2 Chemokine (C-C motif) receptor 2 1.00 26.28 26.17 A10 94Hs,5916 396NM_006139 CD28 CD28 molecule 0.81 34.51 34.71 A11 29Hs,5126 NM_001712 CEACAM1 Carcinoembryonic antigen-related cell adhesion molecule 1 1.31 26.08 25.59 B01 82Hs,2478 NM_005214 CTLA4 (biliaryCytotoxic glycoprotein) T-lymphocyte -associated protein 4 0.53 30.90 31.71 B11 24Hs,208 NM_000160 GCGR Glucagon receptor 0.93 35.00 35.00 C01 Hs,3891 NM_002062 GLP1R Glucagon-like peptide 1 receptor 0.93 35.00 35.00 C07 03Hs,6434 NM_000201 ICAM1 Intercellular adhesion molecule 1 0.84 28.74 28.89 D02 47Hs,5134 NM_000418 IL4R Interleukin 4 receptor 0.64 34.22 34.75 D06 57Hs,4657 NM_000208 INSR Insulin receptor 0.93 35.00 35.00 E05 44Hs,4312 NM_006178 NSF N-ethylmaleimide-sensitive factor 0.48 28.42 29.37 F08 79Hs,2961 NM_004578 RAB4A RAB4A, member RAS oncogene family 0.88 20.55 20.63 F10 69Hs,7287 NM_000655 SELL Selectin L 0.97 23.89 23.83 F11 56Hs,3806 NM_001042 SLC2A4 Solute carrier family 2 (facilitated glucose transporter), member 4 0.77 34.72 35.00 F12 91Hs,5111 NM_003825 SNAP23 Synaptosomal-associated protein, 23kDa 3.90