In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Exonic Splicing Silencer Is Involved in the Regulated Splicing of Glucose 6-Phosphate Dehydrogenase Mrna Wioletta Szeszel-Fedorowicz
Faculty Scholarship 2006 An Exonic Splicing Silencer Is Involved In The Regulated Splicing Of Glucose 6-Phosphate Dehydrogenase Mrna Wioletta Szeszel-Fedorowicz Indrani Talukdar Brian N. Griffith Callee M. Walsh Lisa M. Salati Follow this and additional works at: https://researchrepository.wvu.edu/faculty_publications Digital Commons Citation Szeszel-Fedorowicz, Wioletta; Talukdar, Indrani; Griffith,r B ian N.; Walsh, Callee M.; and Salati, Lisa M., "An Exonic Splicing Silencer Is Involved In The Regulated Splicing Of Glucose 6-Phosphate Dehydrogenase Mrna" (2006). Faculty Scholarship. 464. https://researchrepository.wvu.edu/faculty_publications/464 This Article is brought to you for free and open access by The Research Repository @ WVU. It has been accepted for inclusion in Faculty Scholarship by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 45, pp. 34146–34158, November 10, 2006 © 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A. An Exonic Splicing Silencer Is Involved in the Regulated Splicing of Glucose 6-Phosphate Dehydrogenase mRNA* Received for publication, April 20, 2006, and in revised form, September 14, 2006 Published, JBC Papers in Press, September 15, 2006, DOI 10.1074/jbc.M603825200 Wioletta Szeszel-Fedorowicz, Indrani Talukdar, Brian N. Griffith, Callee M. Walsh, and Lisa M. Salati1 From the Department of Biochemistry and Molecular Pharmacology, West Virginia University, Morgantown, West Virginia 26506 The inhibition of glucose-6-phosphate dehydrogenase (G6PD) in converting excess dietary energy into a storage form. Con- expression by arachidonic acid occurs by changes in the rate sistent with this role in energy homeostasis, the capacity of this of pre-mRNA splicing. -

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
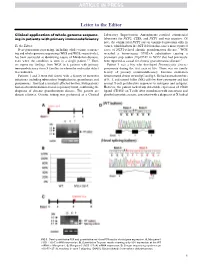
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Analysis of Exonic Mutations Leading to Exon Skipping in Patients with Pyruvate Dehydrogenase E1␣ Deficiency
0031-3998/00/4806-0748 PEDIATRIC RESEARCH Vol. 48, No. 6, 2000 Copyright © 2000 International Pediatric Research Foundation, Inc. Printed in U.S.A. Analysis of Exonic Mutations Leading to Exon Skipping in Patients with Pyruvate Dehydrogenase E1␣ Deficiency ALESSANDRA KUPPER CARDOZO, LINDA DE MEIRLEIR, INGE LIEBAERS, AND WILLY LISSENS Center for Medical Genetics [A.K.C., L.D.M., I.L., W.L.] and Pediatric Neurology [L.D.M.], University Hospital, Vrije Universiteit Brussel, 1090 Brussels, Belgium. ABSTRACT The pyruvate dehydrogenase (PDH) complex is situated at a mutations that either reverted or disrupted the wild-type pre- key position in energy metabolism and is responsible for the dicted pre-mRNA secondary structure of exon 6, we were unable conversion of pyruvate to acetyl CoA. In the literature, two to establish a correlation between the aberrant splicing and unrelated patients with a PDH complex deficiency and splicing disruption of the predicted structure. The mutagenic experiments out of exon 6 of the PDH E1␣ gene have been described, described here and the silent mutation found in one of the although intronic/exonic boundaries on either side of exon 6 patients suggest the presence of an exonic splicing enhancer in were completely normal. Analysis of exon 6 in genomic DNA of the middle region of exon 6 of the PDH E1␣ gene. (Pediatr Res these patients revealed two exonic mutations, a silent and a 48: 748–753, 2000) missense mutation. Although not experimentally demonstrated, the authors in both publications suggested that the exonic muta- tions were responsible for the exon skipping. In this work, we Abbreviations were able to demonstrate, by performing splicing experiments, ESE, exonic splicing enhancer that the two exonic mutations described in the PDH E1␣ gene mfe, minimum free energy lead to aberrant splicing. -

Serine Arginine-Rich Protein-Dependent Suppression Of
Serine͞arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers El Che´ rif Ibrahim*, Thomas D. Schaal†, Klemens J. Hertel‡, Robin Reed*, and Tom Maniatis†§ *Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115; †Department of Molecular and Cellular Biology, Harvard University, 7 Divinity Avenue, Cambridge, MA 02138; and ‡Department of Microbiology and Molecular Genetics, University of California, Irvine, CA 92697-4025 Contributed by Tom Maniatis, January 28, 2005 The 5 and 3 splice sites within an intron can, in principle, be joined mechanisms by which exon skipping is prevented. Our data to those within any other intron during pre-mRNA splicing. How- reveal that splicing to the distal 3Ј splice site is suppressed by ever, exons are joined in a strict 5 to 3 linear order in constitu- proximal exonic sequences and that SR proteins are required for tively spliced pre-mRNAs. Thus, specific mechanisms must exist to this suppression. Thus, SR protein͞exonic enhancer complexes prevent the random joining of exons. Here we report that insertion not only function in exon and splice-site recognition but also play of exon sequences into an intron can inhibit splicing to the a role in ensuring that 5Ј and 3Ј splice sites within the same intron .downstream 3 splice site and that this inhibition is independent of are used, thus suppressing exon skipping intron size. The exon sequences required for splicing inhibition were found to be exonic enhancer elements, and their inhibitory Materials and Methods activity requires the binding of serine͞arginine-rich splicing fac- Construction of Plasmids. -

Position-Dependent Splicing Activation and Repression by SR and Hnrnp Proteins Rely on Common Mechanisms
Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms STEFFEN ERKELENZ,1,3 WILLIAM F. MUELLER,2,3 MELANIE S. EVANS,2 ANKE BUSCH,2 KATRIN SCHO¨ NEWEIS,1 KLEMENS J. HERTEL,2,4 and HEINER SCHAAL1,4 1Institute of Virology, Heinrich-Heine-University, D-40225 Du¨sseldorf, Germany 2Department of Microbiology and Molecular Genetics, University of California, Irvine, Irvine, California 92697-4025, USA ABSTRACT Alternative splicing is regulated by splicing factors that modulate splice site selection. In some cases, however, splicing factors show antagonistic activities by either activating or repressing splicing. Here, we show that these opposing outcomes are based on their binding location relative to regulated 59 splice sites. SR proteins enhance splicing only when they are recruited to the exon. However, they interfere with splicing by simply relocating them to the opposite intronic side of the splice site. hnRNP splicing factors display analogous opposing activities, but in a reversed position dependence. Activation by SR or hnRNP proteins increases splice site recognition at the earliest steps of exon definition, whereas splicing repression promotes the assembly of nonproductive complexes that arrest spliceosome assembly prior to splice site pairing. Thus, SR and hnRNP splicing factors exploit similar mechanisms to positively or negatively influence splice site selection. Keywords: SR protein; hnRNP protein; pre-mRNA splicing; splicing activation; splicing repression INTRODUCTION their RS domain (Fu and Maniatis 1992; Lin and Fu 2007) or through RNA binding domain interactions with U1 Alternative pre-mRNA splicing promotes high proteomic snRNP-specific protein 70K (Cho et al. 2011). However, in diversity despite the limited number of protein coding some cases SR proteins were shown to interfere with exon genes (Nilsen and Graveley 2010). -

Determinants of Exon 7 Splicing in the Spinal Muscular Atrophy Genes, SMN1 and SMN2 Luca Cartegni,*,† Michelle L
Determinants of Exon 7 Splicing in the Spinal Muscular Atrophy Genes, SMN1 and SMN2 Luca Cartegni,*,† Michelle L. Hastings,* John A. Calarco,‡ Elisa de Stanchina, and Adrian R. Krainer Cold Spring Harbor Laboratory, Cold Spring Harbor, NY Spinal muscular atrophy is a neurodegenerative disorder caused by the deletion or mutation of the survival-of- motor-neuron gene, SMN1. An SMN1 paralog, SMN2, differs by a CrT transition in exon 7 that causes substantial skipping of this exon, such that SMN2 expresses only low levels of functional protein. A better understanding of SMN splicing mechanisms should facilitate the development of drugs that increase survival motor neuron (SMN) protein levels by improving SMN2 exon 7 inclusion. In addition, exonic mutations that cause defective splicing give rise to many genetic diseases, and the SMN1/2 system is a useful paradigm for understanding exon-identity determinants and alternative-splicing mechanisms. Skipping of SMN2 exon 7 was previously attributed either to the loss of an SF2/ASF–dependent exonic splicing enhancer or to the creation of an hnRNP A/B–dependent exonic splicing silencer, as a result of the CrT transition. We report the extensive testing of the enhancer-loss and silencer- gain models by mutagenesis, RNA interference, overexpression, RNA splicing, and RNA-protein interaction ex- periments. Our results support the enhancer-loss model but also demonstrate that hnRNP A/B proteins antagonize SF2/ASF–dependent ESE activity and promote exon 7 skipping by a mechanism that is independent of the CrT transition and is, therefore, common to both SMN1 and SMN2. Our findings explain the basis of defective SMN2 splicing, illustrate the fine balance between positive and negative determinants of exon identity and alternative splicing, and underscore the importance of antagonistic splicing factors and exonic elements in a disease context. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Morpholino-Mediated Exon Inclusion for SMA
Morpholino-mediated exon inclusion for SMA Haiyan Zhou1 and Francesco Muntoni1* 1The Dubowitz Neuromuscular Centre, Molecular Neurosciences Session, Developmental Neurosciences Programme, Great Ormond Street Institute of Child Health, University College London. 30 Guilford Street, London WC1N 1EH. United Kingdom. E-mail: [email protected] Abstract The application of antisense oligonucleotides (AONs) to modify pre-messenger RNA splicing has great potential for treating genetic diseases. The strategies used to redirect splicing for therapeutic purpose involve the use of AONs complementary to splice motifs, enhancer or silencer sequences. AONs to block intronic splicing silencer motifs can efficiently augment exon 7 inclusion in survival motor neuron 2 (SMN2) gene and have demonstrated robust therapeutic effects in both pre-clinical studies and clinical trials in spinal muscular atrophy (SMA), which lead to a recently approved drug. AONs with phosphoroamidate morpholino (PMO) backbone have shown target engagement with restoration of the defective protein in Duchenne muscular dystrophy (DMD) and their safety profile lead to a recent conditional approval for one DMD PMO drug. PMO AONs are also effective in correcting SMN2 exon 7 splicing and rescuing SMA transgenic mice. Here we provide the details of methods that our lab has used to evaluate PMO-mediated SMN2 exon 7 inclusion in the in vivo studies conducted in SMA transgenic mice. The methods comprise mouse experiment procedures, assessment of PMOs on exon 7 inclusion at RNA levels by reverse transcription (RT-) PCR and quantitative real-time PCR. In addition, we present methodology for protein quantification using western blot in mouse tissues, on neuropathology assessment of skeletal muscle (muscle pathology and neuromuscular junction staining) as well as behaviour test in the SMA mice (righting reflex). -

Localization of an Exonic Splicing Enhancer Responsible For
3108–3115 Nucleic Acids Research, 2001, Vol. 29, No. 14 © 2001 Oxford University Press Localization of an exonic splicing enhancer responsible for mammalian natural trans-splicing Concha Caudevilla, Carles Codony1, Dolors Serra, Guillem Plasencia, Ruth Román, Adolf Graessmann2, Guillermina Asins, Montserrat Bach-Elias1 and Fausto G. Hegardt* Department of Biochemistry and Molecular Biology, School of Pharmacy, Diagonal 643, University of Barcelona, 08028 Barcelona, Spain, 1IIBB Consejo Superior de Investigaciones Científicas, 08034 Barcelona, Spain and 2Institut für Molekularbiologie, Freie Universität Berlin, D-14195 Berlin, Germany Received February 15, 2001; Revised and Accepted June 1, 2001 DDBJ/EMBL/GenBank accession nos AF144397–AF144399 ABSTRACT recognize exon–intron boundaries at both 5′ and 3′ splice sites and then produce the necessary reactions to ligate the exons Carnitine octanoyltransferase (COT) produces three involved. Two kinds of splicing are observed in nature. different transcripts in rat through cis-andtrans- Cis-splicing, by which introns from the same pre-mRNA are splicing reactions, which may lead to the synthesis removed, and trans-splicing, in which two pre-mRNAs are of two proteins. Generation of the three COT tran- processed to produce a mature transcript that contains exons scripts in rat does not depend on sex, development, from both precursors. The trans-splicing process has been fat feeding, the inclusion of the peroxisome prolifer- described mostly in trypanosome, nematodes, plant/algal ator diethylhexyl phthalate in the diet or hyper- chloroplasts and plant mitochondria (1). insulinemia. In addition, trans-splicing was not A family of serine–arginine-rich RNA binding proteins, detected in COT of other mammals, such as human, known as SR proteins, plays a critical role in the formation of pig, cow and mouse, or in Cos7 cells from monkey.