Expression of Alternative Oxidase in Drosophila Ameliorates Diverse Phenotypes Due to Cytochrome Oxidase Deficiency
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
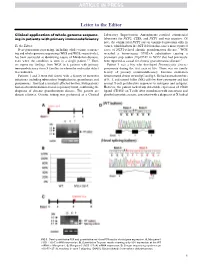
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Tissue-And Condition-Specific Isoforms of Mammalian Cytochrome
Hindawi Oxidative Medicine and Cellular Longevity Volume 2017, Article ID 1534056, 19 pages https://doi.org/10.1155/2017/1534056 Review Article Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease 1 1 1 2 3 Christopher A. Sinkler, Hasini Kalpage, Joseph Shay, Icksoo Lee, Moh H. Malek, 1 1 Lawrence I. Grossman, and Maik Hüttemann 1Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI 48201, USA 2College of Medicine, Dankook University, Cheonan-si, Chungcheongnam-do 31116, Republic of Korea 3Department of Health Care Sciences, Eugene Applebaum College of Pharmacy and Health Sciences, Wayne State University, Detroit, MI 48201, USA Correspondence should be addressed to Maik Hüttemann; [email protected] Received 2 February 2017; Accepted 29 March 2017; Published 16 May 2017 Academic Editor: Ryuichi Morishita Copyright © 2017 Christopher A. Sinkler et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Cytochrome c oxidase (COX) is the terminal enzyme of the electron transport chain and catalyzes the transfer of electrons from cytochrome c to oxygen. COX consists of 14 subunits, three and eleven encoded, respectively, by the mitochondrial and nuclear DNA. Tissue- and condition-specific isoforms have only been reported for COX but not for the other oxidative phosphorylation complexes, suggesting a fundamental requirement to fine-tune and regulate the essentially irreversible reaction catalyzed by COX. This article briefly discusses the assembly of COX in mammals and then reviews the functions of the six nuclear-encoded COX subunits that are expressed as isoforms in specialized tissues including those of the liver, heart and skeletal muscle, lung, and testes: COX IV-1, COX IV-2, NDUFA4, NDUFA4L2, COX VIaL, COX VIaH, COX VIb-1, COX VIb-2, COX VIIaH, COX VIIaL, COX VIIaR, COX VIIIH/L, and COX VIII-3. -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -

A Mutational Analysis of Spliceosome Assembly: Evidence for Splice Site Collaboration During Spliceosome Formation
Downloaded from genesdev.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press A mutational analysis of spliceosome assembly: evidence for splice site collaboration during spliceosome formation Angus I. Lamond, Maria M. Konarska, and Phillip A. Sharp Center for Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139 USA We have analyzed the pathway of mammalian spliceosome assembly in vitro using a mobility retardation assay. The binding of splicing complexes to both wild-type and mutant [3-globin pre-RNAs was studied. Three kinetically related, ATP-dependent complexes, a, ~, and % were resolved with a wild-type [3-globin substrate. These complexes formed, both temporally and in order of decreasing mobility, ~ -, [3 --, ~/. All three complexes contained U2 snRNA. The RNA intermediates of splicing, i.e., free 5' exon and intron lariat + 3' exon, were found predominantly in the ~/complex. The RNA products of splicing, i.e., ligated exons and fully excised intron lariat, were found in separate, postsplicing complexes which appeared to form via breakdown of ~/. Mutations of the 5' splice site, which caused an accumulation of splicing intermediates, also resulted in accumulation of the ~/complex. Mutations of the 3' splice site, which severely inhibited splicing, reduced the efficiency and altered the pattern of complex formation. Surprisingly, the analysis of double mutants, with sequence alterations at both the 5' and 3' splice sites, revealed that the 5' splice site genotype was important for the efficient formation of a U2 snRNA-containing a complex at the 3' splice site. Thus, it appears that a collaborative interaction between the separate 5' and 3' splice sites promotes spliceosome assembly. -

Genomics of Inherited Bone Marrow Failure and Myelodysplasia Michael
Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2015 Reading Committee: Mary-Claire King, Chair Akiko Shimamura Marshall Horwitz Program Authorized to Offer Degree: Molecular and Cellular Biology 1 ©Copyright 2015 Michael Yu Zhang 2 University of Washington ABSTRACT Genomics of inherited bone marrow failure and myelodysplasia Michael Yu Zhang Chair of the Supervisory Committee: Professor Mary-Claire King Department of Medicine (Medical Genetics) and Genome Sciences Bone marrow failure and myelodysplastic syndromes (BMF/MDS) are disorders of impaired blood cell production with increased leukemia risk. BMF/MDS may be acquired or inherited, a distinction critical for treatment selection. Currently, diagnosis of these inherited syndromes is based on clinical history, family history, and laboratory studies, which directs the ordering of genetic tests on a gene-by-gene basis. However, despite extensive clinical workup and serial genetic testing, many cases remain unexplained. We sought to define the genetic etiology and pathophysiology of unclassified bone marrow failure and myelodysplastic syndromes. First, to determine the extent to which patients remained undiagnosed due to atypical or cryptic presentations of known inherited BMF/MDS, we developed a massively-parallel, next- generation DNA sequencing assay to simultaneously screen for mutations in 85 BMF/MDS genes. Querying 71 pediatric and adult patients with unclassified BMF/MDS using this assay revealed 8 (11%) patients with constitutional, pathogenic mutations in GATA2 , RUNX1 , DKC1 , or LIG4 . All eight patients lacked classic features or laboratory findings for their syndromes. -

The Ribose Methylation Enzyme FTSJ1 Has a Conserved Role in Neuron Morphology and Learning Performance
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.06.430044; this version posted July 25, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The ribose methylation enzyme FTSJ1 has a conserved role in neuron morphology and learning performance Dilyana G Dimitrova1*, Mira Brazane1*, Julien Pigeon2, Chiara Paolantoni3, Tao Ye4, Virginie Marchand5, Bruno Da Silva1, Elise Schaefer6, Margarita T Angelova1, Zornitza Stark7, Martin Delatycki7, Tracy Dudding-Byth8, Jozef Gecz9, Pierre-Yves Placais10, Laure Teysset1, Thomas Preat10, Amélie Piton4, Bassem A. Hassan2, Jean-Yves Roignant3,11, Yuri Motorin12 and Clément Carré1,#. 1Transgenerational Epigenetics & small RNA Biology, Sorbonne Université, Centre National de la Recherche Scientifique, Laboratoire de Biologie du Développement - Institut de Biologie Paris Seine, 9 Quai Saint Bernard, 75005 Paris, France. 2Paris Brain Institute-Institut du Cerveau (ICM), Sorbonne Université, Inserm, CNRS, Hôpital Pitié-Salpêtrière, Paris, France. 3Center for Integrative Genomics, Génopode Building, Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland. 4Institute of Genetics and Molecular and Cellular Biology, Strasbourg University, CNRS UMR7104, INSERM U1258, 67400 Illkirch, France. 5Université de Lorraine, CNRS, INSERM, EpiRNASeq Core Facility, UMS2008/US40 IBSLor ,F-54000 Nancy, France. 6Service de Génétique Médicale, Hôpitaux Universitaires de Strasbourg, Institut de Génétique Médicale d’Alsace, Strasbourg, France. 7Victorian Clinical Genetics Services, Murdoch Children's Research Institute, Melbourne, VIC, Australia. Department of Paediatrics, The University of Melbourne, Melbourne, VIC, Australia. 8University of Newcastle, Newcastle, NSW Australia. -

Diagnostic Interpretation of Genetic Studies in Patients with Primary
AAAAI Work Group Report Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology Ivan K. Chinn, MD,a,b Alice Y. Chan, MD, PhD,c Karin Chen, MD,d Janet Chou, MD,e,f Morna J. Dorsey, MD, MMSc,c Joud Hajjar, MD, MS,a,b Artemio M. Jongco III, MPH, MD, PhD,g,h,i Michael D. Keller, MD,j Lisa J. Kobrynski, MD, MPH,k Attila Kumanovics, MD,l Monica G. Lawrence, MD,m Jennifer W. Leiding, MD,n,o,p Patricia L. Lugar, MD,q Jordan S. Orange, MD, PhD,r,s Kiran Patel, MD,k Craig D. Platt, MD, PhD,e,f Jennifer M. Puck, MD,c Nikita Raje, MD,t,u Neil Romberg, MD,v,w Maria A. Slack, MD,x,y Kathleen E. Sullivan, MD, PhD,v,w Teresa K. Tarrant, MD,z Troy R. Torgerson, MD, PhD,aa,bb and Jolan E. Walter, MD, PhDn,o,cc Houston, Tex; San Francisco, Calif; Salt Lake City, Utah; Boston, Mass; Great Neck and Rochester, NY; Washington, DC; Atlanta, Ga; Rochester, Minn; Charlottesville, Va; St Petersburg, Fla; Durham, NC; Kansas City, Mo; Philadelphia, Pa; and Seattle, Wash AAAAI Position Statements,Work Group Reports, and Systematic Reviews are not to be considered to reflect current AAAAI standards or policy after five years from the date of publication. The statement below is not to be construed as dictating an exclusive course of action nor is it intended to replace the medical judgment of healthcare professionals.