Impaired Hematopoiesis and Leukemia Development in Mice with a Conditional Knock-In Allele of a Mutant Splicing Factor Gene U2af1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
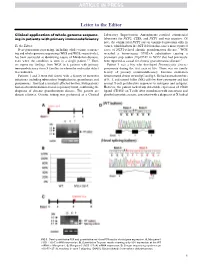
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Prognostic Biomarkers in Myelodysplastic Syndromes
Myelodysplastic syndromes Prognostic biomarkers in myelodysplastic syndromes M. Cazzola ABSTRACT Prognostic biomarkers in myelodysplastic syndromes (MDS) include cytogenetic abnormalities and Department of Hematology somatic gene mutations. Recurrent chromosomal abnormalities are found in approximately 50% of Oncology, Fondazione Istituto cases, and are mainly secondary genetic events. By contrast, somatic oncogenic mutations, responsible di Ricovero e Cura a Carattere for disease pathogenesis and progression, are found in up to 90% of MDS patients. Oncogenic muta - Scientifico (IRCCS) Policlinico tions can be classified as: i) founding or initiating driver mutations, which cause a selective advantage San Matteo, and Department in a hematopoietic cell with capacity for self-renewal and lead to formation of a clone of mutated of Molecular Medicine, myelodysplastic cells; ii) subclonal or cooperating driver mutations, which occur in cells of an already University of Pavia, Pavia, Italy established clone and generate subclones carrying both the founding and the newly acquired muta - tion. Driver mutant genes include those of RNA splicing, DNA methylation, histone modification, tran - Correspondence: Mario Cazzola scription regulation, DNA repair, signal transduction, and cohesin complex. Only 6 genes ( TET2, SF3B1, E-mail: [email protected] ASXL1, SRSF2, DNMT3A , and RUNX1 ) are consistently mutated in 10% or more of MDS patients, while Acknowledgments: a long tail of additional genes are mutated less frequently. Reliable genotype/phenotype relationships The studies on the genetic basis of have already been established, primarily the close association between SF3B1 mutation and ring sider - myeloid neoplasms conducted at the oblasts. Clonal and subclonal mutations appear to affect prognosis equally, and outcome correlates Department of Hematology with the number of driver mutations. -

Aiolos Overexpression in Systemic Lupus Erythematosus B Cell
Aiolos Overexpression in Systemic Lupus Erythematosus B Cell Subtypes and BAFF-Induced Memory B Cell Differentiation Are Reduced by CC-220 This information is current as Modulation of Cereblon Activity of September 27, 2021. Yumi Nakayama, Jolanta Kosek, Lori Capone, Eun Mi Hur, Peter H. Schafer and Garth E. Ringheim J Immunol 2017; 199:2388-2407; Prepublished online 28 August 2017; Downloaded from doi: 10.4049/jimmunol.1601725 http://www.jimmunol.org/content/199/7/2388 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2017/08/26/jimmunol.160172 Material 5.DCSupplemental References This article cites 131 articles, 45 of which you can access for free at: http://www.jimmunol.org/content/199/7/2388.full#ref-list-1 Why The JI? Submit online. by guest on September 27, 2021 • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Author Choice Freely available online through The Journal of Immunology Author Choice option Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

Detection of Pathogenic Isoforms of IKZF1 in Leukemic Cell
cancers Article Detection of Pathogenic Isoforms of IKZF1 in Leukemic Cell Lines and Acute Lymphoblastic Leukemia Samples: Identification of a Novel Truncated IKZF1 Transcript in SUP-B15 Weiqiang Zhao 1,*, Ying Li 2 , Chenjiao Yao 2 , Guojuan Zhang 1, Kevin Y. Zhao 1, Wei Chen 1, Peng Ru 1, Xiaokang Pan 1, Huolin Tu 1 and Daniel Jones 1 1 The James Comprehensive Cancer Center and Solove Research Institute, The Ohio State University, Columbus, OH 43210, USA; [email protected] (G.Z.); [email protected] (K.Y.Z.); [email protected] (W.C.); [email protected] (P.R.); [email protected] (X.P.); [email protected] (H.T.); [email protected] (D.J.) 2 The Department of Pediatrics and Department of Hematology, Xiang-Ya Third Hospital, Changsha 410013, China; [email protected] (Y.L.); [email protected] (C.Y.) * Correspondence: [email protected]; Tel.: +1-6142934210 Received: 27 August 2020; Accepted: 22 October 2020; Published: 28 October 2020 Simple Summary: Abnormal RNA splicing plays a fundamental role in leukemogenesis in acute lymphoblastic leukemia (ALL). Many cases of high-risk B-cell ALL cases, including BCR-ABL1+ and BCR-ABL1-like ALL, share a common molecular mechanism of aberrant fusion transcripts involving tyrosine kinase genes combined with dysregulation of the transcription factor and lymphocyte differentiation factor IKZF1. Dysfunction of IKZF1 in ALL is caused by mutation and gene deletion but also alternative splicing resulting in exon skipping with production of aberrant IKZF1 proteins. We report here an assay to detect aberrantly spliced isoforms of IKZF1 in ALL to assist in diagnosis, outcome prediction, and therapy selection in ALL and the identification of a novel altered IKZF1 product in a model ALL cell line. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Novel Gene Targets Detected by Genomic Profiling in a Consecutive Series of 126 Adults with Acute Lymphoblastic Leukemia
Acute Lymphoblastic Leukemia ARTICLES Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia Setareh Safavi,1 Markus Hansson,2 Karin Karlsson,2 Andrea Biloglav,1 Bertil Johansson,1,3 and Kajsa Paulsson1 1Division of Clinical Genetics, Department of Laboratory Medicine, Lund University; 2Division of Hematology, Skåne University Hospital, Lund University; and 3Department of Clinical Genetics, University and Regional Laboratories, Region Skåne, Lund, Sweden ABSTRACT In contrast to acute lymphoblastic leukemia in children, adult cases of this disease are associated with a very poor prognosis. In order to ascertain whether the frequencies and patterns of submicroscopic changes, identifiable with single nucleotide polymorphism array analysis, differ between childhood and adult acute lymphoblastic leukemia, we performed single nucleotide polymorphism array analyses of 126 adult cases, the largest series to date, includ- ing 18 paired diagnostic and relapse samples. Apart from identifying characteristic microdeletions of the CDKN2A, EBF1, ETV6, IKZF1, PAX5 and RB1 genes, the present study uncovered novel, focal deletions of the BCAT1, BTLA, NR3C1, PIK3AP1 and SERP2 genes in 2-6% of the adult cases. IKZF1 deletions were associated with B-cell pre- cursor acute lymphoblastic leukemia (P=0.036), BCR-ABL1-positive acute lymphoblastic leukemia (P<0.001), and higher white blood cell counts (P=0.005). In addition, recurrent deletions of RASSF3 and TOX were seen in relapse samples. Comparing paired diagnostic/relapse samples revealed identical changes at diagnosis and relapse in 27%, clonal evolution in 22%, and relapses evolving from ancestral clones in 50%, akin to what has previously been reported in pediatric acute lymphoblastic leukemia and indicating that the mechanisms of relapse may be similar in adult and childhood cases. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Immunogenic Dendritic Cell Generation from Pluripotent Stem Cells by Ectopic Expression of Runx3
Immunogenic Dendritic Cell Generation from Pluripotent Stem Cells by Ectopic Expression of Runx3 This information is current as Erika Takacs, Pal Boto, Emilia Simo, Tamas I. Csuth, of September 25, 2021. Bianka M. Toth, Hadas Raveh-Amit, Attila Pap, Elek G. Kovács, Julianna Kobolak, Szilvia Benkö, Andras Dinnyes and Istvan Szatmari J Immunol published online 16 November 2016 http://www.jimmunol.org/content/early/2016/11/15/jimmun Downloaded from ol.1600034 Supplementary http://www.jimmunol.org/content/suppl/2016/11/15/jimmunol.160003 Material 4.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 25, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published November 16, 2016, doi:10.4049/jimmunol.1600034 The Journal of Immunology Immunogenic Dendritic Cell Generation from Pluripotent Stem Cells by Ectopic Expression of Runx3 Erika Takacs,*,1 Pal Boto,*,1 Emilia Simo,* Tamas I. Csuth,* Bianka M.