REVIEW Novel Mutations Associated with Combined Pituitary Hormone
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -

Activated Peripheral-Blood-Derived Mononuclear Cells
Transcription factor expression in lipopolysaccharide- activated peripheral-blood-derived mononuclear cells Jared C. Roach*†, Kelly D. Smith*‡, Katie L. Strobe*, Stephanie M. Nissen*, Christian D. Haudenschild§, Daixing Zhou§, Thomas J. Vasicek¶, G. A. Heldʈ, Gustavo A. Stolovitzkyʈ, Leroy E. Hood*†, and Alan Aderem* *Institute for Systems Biology, 1441 North 34th Street, Seattle, WA 98103; ‡Department of Pathology, University of Washington, Seattle, WA 98195; §Illumina, 25861 Industrial Boulevard, Hayward, CA 94545; ¶Medtronic, 710 Medtronic Parkway, Minneapolis, MN 55432; and ʈIBM Computational Biology Center, P.O. Box 218, Yorktown Heights, NY 10598 Contributed by Leroy E. Hood, August 21, 2007 (sent for review January 7, 2007) Transcription factors play a key role in integrating and modulating system. In this model system, we activated peripheral-blood-derived biological information. In this study, we comprehensively measured mononuclear cells, which can be loosely termed ‘‘macrophages,’’ the changing abundances of mRNAs over a time course of activation with lipopolysaccharide (LPS). We focused on the precise mea- of human peripheral-blood-derived mononuclear cells (‘‘macro- surement of mRNA concentrations. There is currently no high- phages’’) with lipopolysaccharide. Global and dynamic analysis of throughput technology that can precisely and sensitively measure all transcription factors in response to a physiological stimulus has yet to mRNAs in a system, although such technologies are likely to be be achieved in a human system, and our efforts significantly available in the near future. To demonstrate the potential utility of advanced this goal. We used multiple global high-throughput tech- such technologies, and to motivate their development and encour- nologies for measuring mRNA levels, including massively parallel age their use, we produced data from a combination of two distinct signature sequencing and GeneChip microarrays. -
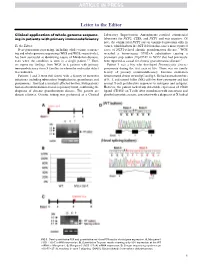
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Table 2. Significant
Table 2. Significant (Q < 0.05 and |d | > 0.5) transcripts from the meta-analysis Gene Chr Mb Gene Name Affy ProbeSet cDNA_IDs d HAP/LAP d HAP/LAP d d IS Average d Ztest P values Q-value Symbol ID (study #5) 1 2 STS B2m 2 122 beta-2 microglobulin 1452428_a_at AI848245 1.75334941 4 3.2 4 3.2316485 1.07398E-09 5.69E-08 Man2b1 8 84.4 mannosidase 2, alpha B1 1416340_a_at H4049B01 3.75722111 3.87309653 2.1 1.6 2.84852656 5.32443E-07 1.58E-05 1110032A03Rik 9 50.9 RIKEN cDNA 1110032A03 gene 1417211_a_at H4035E05 4 1.66015788 4 1.7 2.82772795 2.94266E-05 0.000527 NA 9 48.5 --- 1456111_at 3.43701477 1.85785922 4 2 2.8237185 9.97969E-08 3.48E-06 Scn4b 9 45.3 Sodium channel, type IV, beta 1434008_at AI844796 3.79536664 1.63774235 3.3 2.3 2.75319499 1.48057E-08 6.21E-07 polypeptide Gadd45gip1 8 84.1 RIKEN cDNA 2310040G17 gene 1417619_at 4 3.38875643 1.4 2 2.69163229 8.84279E-06 0.0001904 BC056474 15 12.1 Mus musculus cDNA clone 1424117_at H3030A06 3.95752801 2.42838452 1.9 2.2 2.62132809 1.3344E-08 5.66E-07 MGC:67360 IMAGE:6823629, complete cds NA 4 153 guanine nucleotide binding protein, 1454696_at -3.46081884 -4 -1.3 -1.6 -2.6026947 8.58458E-05 0.0012617 beta 1 Gnb1 4 153 guanine nucleotide binding protein, 1417432_a_at H3094D02 -3.13334396 -4 -1.6 -1.7 -2.5946297 1.04542E-05 0.0002202 beta 1 Gadd45gip1 8 84.1 RAD23a homolog (S. -

Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins
International Journal of Molecular Sciences Article Bioinformatic Analysis of Structure and Function of LIM Domains of Human Zyxin Family Proteins M. Quadir Siddiqui 1,† , Maulik D. Badmalia 1,† and Trushar R. Patel 1,2,3,* 1 Alberta RNA Research and Training Institute, Department of Chemistry and Biochemistry, University of Lethbridge, 4401 University Drive, Lethbridge, AB T1K 3M4, Canada; [email protected] (M.Q.S.); [email protected] (M.D.B.) 2 Department of Microbiology, Immunology and Infectious Disease, Cumming School of Medicine, University of Calgary, 3330 Hospital Drive, Calgary, AB T2N 4N1, Canada 3 Li Ka Shing Institute of Virology, University of Alberta, Edmonton, AB T6G 2E1, Canada * Correspondence: [email protected] † These authors contributed equally to the work. Abstract: Members of the human Zyxin family are LIM domain-containing proteins that perform critical cellular functions and are indispensable for cellular integrity. Despite their importance, not much is known about their structure, functions, interactions and dynamics. To provide insights into these, we used a set of in-silico tools and databases and analyzed their amino acid sequence, phylogeny, post-translational modifications, structure-dynamics, molecular interactions, and func- tions. Our analysis revealed that zyxin members are ohnologs. Presence of a conserved nuclear export signal composed of LxxLxL/LxxxLxL consensus sequence, as well as a possible nuclear localization signal, suggesting that Zyxin family members may have nuclear and cytoplasmic roles. The molecular modeling and structural analysis indicated that Zyxin family LIM domains share Citation: Siddiqui, M.Q.; Badmalia, similarities with transcriptional regulators and have positively charged electrostatic patches, which M.D.; Patel, T.R. -

Specificity Landscapes Unmask Submaximal Binding Site Preferences of Transcription Factors
Specificity landscapes unmask submaximal binding site preferences of transcription factors Devesh Bhimsariaa,b,1, José A. Rodríguez-Martíneza,2, Junkun Panc, Daniel Rostonc, Elif Nihal Korkmazc, Qiang Cuic,3, Parameswaran Ramanathanb, and Aseem Z. Ansaria,d,4 aDepartment of Biochemistry, University of Wisconsin–Madison, Madison, WI 53706; bDepartment of Electrical and Computer Engineering, University of Wisconsin–Madison, Madison, WI 53706; cDepartment of Chemistry, University of Wisconsin–Madison, Madison, WI 53706; and dThe Genome Center of Wisconsin, University of Wisconsin–Madison, Madison, WI 53706 Edited by Michael Levine, Princeton University, Princeton, NJ, and approved September 24, 2018 (received for review July 13, 2018) We have developed Differential Specificity and Energy Landscape Here, we report the development of Differential Specificity (DiSEL) analysis to comprehensively compare DNA–protein inter- and Energy Landscapes (DiSEL) to compare experimental actomes (DPIs) obtained by high-throughput experimental plat- platforms, computational methods, and interactomes of TFs, forms and cutting edge computational methods. While high-affinity especially those factors that bind identical consensus motifs. Our DNA binding sites are identified by most methods, DiSEL uncovered results reveal that (i) most high-throughput experimental plat- nuanced sequence preferences displayed by homologous transcription forms reliably identify high-affinity motifs but yield less reliable factors. Pairwise analysis of 726 DPIs uncovered homolog-specific dif- information on submaximal sites; (ii) with few exceptions, com- ferences at moderate- to low-affinity binding sites (submaximal sites). putational methods model DPIs with a focus on high-affinity DiSEL analysis of variants of 41 transcription factors revealed that sites; (iii) submaximal sites improve the annotation of biologi- many disease-causing mutations result in allele-specific changes in cally relevant binding sites across genomes; (iv) among members binding site preferences. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name LIM homeobox 4 Gene Symbol LHX4 Organism Human Gene Summary This gene encodes a member of a large protein family which contains the LIM domain a unique cysteine-rich zinc-binding domain. The encoded protein is a transcription factor involved in the control of differentiation and development of the pituitary gland. Mutations in this gene cause combined pituitary hormone deficiency 4. Gene Aliases CPHD4, FLJ22769 RefSeq Accession No. NC_000001.10, NG_008081.1, NT_004487.19 UniGene ID Hs.658487 Ensembl Gene ID ENSG00000121454 Entrez Gene ID 89884 Assay Information Unique Assay ID qHsaCED0002909 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000263726 Amplicon Context Sequence CTATGGAATCCCCCAGTCTCCATCCTCCATATCGTCCCTGCCATCCCACGCTCCT TTGCTCAATGGGCTGGATTACACGGTGGACAGTAATTTGGGCATCATTGC Amplicon Length (bp) 75 Chromosome Location 1:180243462-180243566 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 94 R2 0.9993 cDNA Cq 24.14 cDNA Tm (Celsius) 82.5 gDNA Cq 23.49 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report LHX4, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

MECHANISMS in ENDOCRINOLOGY: Novel Genetic Causes of Short Stature
J M Wit and others Genetics of short stature 174:4 R145–R173 Review MECHANISMS IN ENDOCRINOLOGY Novel genetic causes of short stature 1 1 2 2 Jan M Wit , Wilma Oostdijk , Monique Losekoot , Hermine A van Duyvenvoorde , Correspondence Claudia A L Ruivenkamp2 and Sarina G Kant2 should be addressed to J M Wit Departments of 1Paediatrics and 2Clinical Genetics, Leiden University Medical Center, PO Box 9600, 2300 RC Leiden, Email The Netherlands [email protected] Abstract The fast technological development, particularly single nucleotide polymorphism array, array-comparative genomic hybridization, and whole exome sequencing, has led to the discovery of many novel genetic causes of growth failure. In this review we discuss a selection of these, according to a diagnostic classification centred on the epiphyseal growth plate. We successively discuss disorders in hormone signalling, paracrine factors, matrix molecules, intracellular pathways, and fundamental cellular processes, followed by chromosomal aberrations including copy number variants (CNVs) and imprinting disorders associated with short stature. Many novel causes of GH deficiency (GHD) as part of combined pituitary hormone deficiency have been uncovered. The most frequent genetic causes of isolated GHD are GH1 and GHRHR defects, but several novel causes have recently been found, such as GHSR, RNPC3, and IFT172 mutations. Besides well-defined causes of GH insensitivity (GHR, STAT5B, IGFALS, IGF1 defects), disorders of NFkB signalling, STAT3 and IGF2 have recently been discovered. Heterozygous IGF1R defects are a relatively frequent cause of prenatal and postnatal growth retardation. TRHA mutations cause a syndromic form of short stature with elevated T3/T4 ratio. Disorders of signalling of various paracrine factors (FGFs, BMPs, WNTs, PTHrP/IHH, and CNP/NPR2) or genetic defects affecting cartilage extracellular matrix usually cause disproportionate short stature. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up,