Spliceosomal Pre-Mrna Splicing Methods and Protocols M ETHODS in MOLECULAR BIOLOGY
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Jpn. J. Protozool. 38(2) 171-183
Jpn. J. Protozool. Vol. 38, No. 2. (2005) 171 Review On the origin of mitochondria and Rickettsia-related eukaryotic endo- symbionts B. Franz Lang1*, Henner Brinkmann1, Liisa B. Koski1, Masahiro Fujishima2, Hans-Dieter Görtz3 and Gertraud Burger1 1Program in Evolutionary Biology, Canadian Institute for Advanced Research; Centre Robert Cedergren, Département de Biochimie, Université de Montréal, 2900 Boulevard Edouard- Montpetit, Montréal, Québec, H3T 1J4, Canada. 2Biological Institute, Faculty of Science, Yamaguchi University, Yamaguchi 753-8512, Japan. 3Abteilung Zoologie, Biologisches Institut, Universität Stuttgart, Pfaffenwaldring 57, 70550 Stuttgart, Germany. SUMMARY sence of genes for oxidative phosphorylation, the TCA cycle, and many other metabolic pathways, Resent insights into the origin and early evo- but the presence of several pathogenesis-related lution of mitochondria come from two approaches: genes and a high number of bacterial IS elements. the investigation of mtDNAs from minimally de- Phylogenetic analyses with multiple protein se- rived (primitive) mitochondriate eukaryotes, in quences place H. obtusa basally to the Rickettsia- particular jakobid flagellates, and of genomes from Ehrlichia-Wolbachia assemblage of bacterial intracellular α-proteobacterial symbionts. Of par- pathogens. This leads us to postulate that H. ob- ticular interest in this context is Holospora obtusa, tusa is the closest bacterial relative of mitochon- an intracellular bacterial endosymbiont that resides dria known to date. and replicates in the somatic nucleus of its eu- karyotic host, the ciliate Paramecium caudatum. Currently we have sequenced close to 50% of the INTRODUCTION ~ 1.7 Mbp H. obtusa genome, revealing the ab- One of the major advancements in under- standing eukaryotic evolution was the discovery that mitochondria evolved from an endosymbiotic α-Proteobacterium, and that mitochondrial DNA *Corresponding author (mtDNA) is a relict bacterial genome. -

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
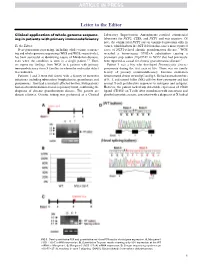
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Sex Is a Ubiquitous, Ancient, and Inherent Attribute of Eukaryotic Life
PAPER Sex is a ubiquitous, ancient, and inherent attribute of COLLOQUIUM eukaryotic life Dave Speijera,1, Julius Lukešb,c, and Marek Eliášd,1 aDepartment of Medical Biochemistry, Academic Medical Center, University of Amsterdam, 1105 AZ, Amsterdam, The Netherlands; bInstitute of Parasitology, Biology Centre, Czech Academy of Sciences, and Faculty of Sciences, University of South Bohemia, 370 05 Ceské Budejovice, Czech Republic; cCanadian Institute for Advanced Research, Toronto, ON, Canada M5G 1Z8; and dDepartment of Biology and Ecology, University of Ostrava, 710 00 Ostrava, Czech Republic Edited by John C. Avise, University of California, Irvine, CA, and approved April 8, 2015 (received for review February 14, 2015) Sexual reproduction and clonality in eukaryotes are mostly Sex in Eukaryotic Microorganisms: More Voyeurs Needed seen as exclusive, the latter being rather exceptional. This view Whereas absence of sex is considered as something scandalous for might be biased by focusing almost exclusively on metazoans. a zoologist, scientists studying protists, which represent the ma- We analyze and discuss reproduction in the context of extant jority of extant eukaryotic diversity (2), are much more ready to eukaryotic diversity, paying special attention to protists. We accept that a particular eukaryotic group has not shown any evi- present results of phylogenetically extended searches for ho- dence of sexual processes. Although sex is very well documented mologs of two proteins functioning in cell and nuclear fusion, in many protist groups, and members of some taxa, such as ciliates respectively (HAP2 and GEX1), providing indirect evidence for (Alveolata), diatoms (Stramenopiles), or green algae (Chlor- these processes in several eukaryotic lineages where sex has oplastida), even serve as models to study various aspects of sex- – not been observed yet. -

BMC Research Notes Biomed Central
BMC Research Notes BioMed Central Short Report Open Access A split and rearranged nuclear gene encoding the iron-sulfur subunit of mitochondrial succinate dehydrogenase in Euglenozoa Ryan MR Gawryluk and Michael W Gray* Address: Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia B3H 1X5, Canada Email: Ryan MR Gawryluk - [email protected]; Michael W Gray* - [email protected] * Corresponding author Published: 3 February 2009 Received: 18 December 2008 Accepted: 3 February 2009 BMC Research Notes 2009, 2:16 doi:10.1186/1756-0500-2-16 This article is available from: http://www.biomedcentral.com/1756-0500/2/16 © 2009 Gray et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Analyses based on phylogenetic and ultrastructural data have suggested that euglenids (such as Euglena gracilis), trypanosomatids and diplonemids are members of a monophyletic lineage termed Euglenozoa. However, many uncertainties are associated with phylogenetic reconstructions for ancient and rapidly evolving groups; thus, rare genomic characters become increasingly important in reinforcing inferred phylogenetic relationships. Findings: We discovered that the iron-sulfur subunit (SdhB) of mitochondrial succinate dehydrogenase is encoded by a split and rearranged nuclear gene in Euglena gracilis and trypanosomatids, an example of a rare genomic character. The two subgenic modules are transcribed independently and the resulting mRNAs appear to be independently translated, with the two protein products imported into mitochondria, based on the presence of predicted mitochondrial targeting peptides. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

News & Views Research
NEWS & VIEWS RESEARCH may exhibit some tissue specificity in humans. Upstream intron Downstream intron a Sibley et al. found that genes with long introns sequence sequence tend to be expressed in the human nervous Precursor RNA system, and they identified recursively spliced 3' Splice 5' Splice RNAs expressed in the human brain6. Duff site site First step et al. detected some selectivity for recursive splicing in the brain in a screen of 20 human tissues (including fetal brain and adult cerebel- Second step lum), but this may partly reflect the difficulty of detecting recursively spliced RNAs in tis- Mature mRNA sues that express such RNAs at low levels. It will be important to determine whether this b RS exon specificity, if real, results from the tendency of recursively spliced genes to be expressed in the brain, or whether cells in the nervous system have factors that promote recursive Competing 5' splice sites First step splicing. Many genes that have long introns, including those that undergo recursive splicing, are linked to neurological diseases and to Second step 9–11 Second step autism . Whether these conditions are sometimes triggered by errors in the multi- step recursive RNA-splicing process will be an exciting avenue for future studies. ■ NMD Heidi Cook-Andersen and Miles F. Wilkinson are in the Department of Reproductive Medicine, University of Figure 1 | Mechanisms of recursive splicing. a, In recursive splicing, long intron sequences of precursor California, San Diego, La Jolla, California, RNA are removed in a stepwise process mediated by juxtaposed internal 3ʹ and 5ʹ splice sites. In the first step, 92093, USA. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

(Bsc Zoology and Microbiology) Concept of Introns and Exons
Unit-5 Molecular Biology (BSc Zoology and Microbiology) Concept of introns and exons Most of the portion of a gene in higher eukaryotes consists of noncoding DNA that interrupts the relatively short segments of coding DNA. The coding sequences are called exons. The noncoding sequences are called introns. Intron: An intron is a portion of a gene that does not code for amino acids An intron is any nucleotide sequence within a gene which is represented in the primary transcript of the gene, but not present in the final processed form. In other words, Introns are noncoding regions of an RNA transcript which are eliminated by splicing before translation. Sequences that are joined together in the final mature RNA after RNA splicing are exons. Introns are very large chunks of RNA within a messenger RNA molecule that interfere with the code of the exons. And these introns get removed from the RNA molecule to leave a string of exons attached to each other so that the appropriate amino acids can be encoded for. Introns are rare in genes of prokaryotes. #Look carefully at the diagram above, we have already discussed about the modification and processing of eukaryotic RNA. In which 5’ guanine cap and 3’poly A tail is added. So at that time, noncoding regions i.e. introns are removed. We hv done ths already. Ok Exon: The coding sequences are called Exon. An exon is the portion of a gene that codes for amino acids. In the cells of plants and animals, most gene sequences are broken up by one or more DNA sequences called introns. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Current Perspectives in Intronic Micro Rnas (Mirnas)
Journal of Biomedical Science (2006) 13:5–15 5 DOI 10.1007/s11373-005-9036-8 Current perspectives in intronic micro RNAs (miRNAs) Shao-Yao Ying & Shi-Lung Lin Department of Cell & Neurobiology, Keck School of Medicine, BMT-403, University of Southern California, 1333 San Pablo Street, Los Angeles, CA, 90033, USA Received 27 May 2005; accepted 14 September 2005 Ó 2005 National Science Council, Taipei Key words: fine-tuning of gene function, functional/structural genomics, gene expression, genetic regula- tion, intronic microRNA, miRNA biogenesis, miRNA, post-translational modification, regulatory gene Summary MicroRNAs (miRNAs), small single-stranded regulatory RNAs capable of interfering with intracellular messenger RNAs (mRNAs) that contain either complete or partial complementarity, are useful for the design of new therapies against cancer polymorphism and viral mutation. Numerous miRNAs have been reported to induce RNA interference (RNAi), a post-transcriptional gene silencing mechanism. Intronic miRNAs, derived from introns by RNA splicing and Dicer processing, can interfere with intracellular mRNAs to silence that gene expression. The intronic miRNAs differ uniquely from previously described intergenic miRNAs in the requirement of type II RNA polymerases (Pol-II) and spliceosomal components for its biogenesis. Several kinds of intronic miRNAs have been identified in Caenorhabditis elegans, mouse and human cells; however, neither their function nor application has been reported. To this day, the computer searching program for miRNA seldom include the intronic portion of protein-coding RNAs. The functional significance of artificially generated intronic miRNAs has been successfully ascertained in several biological systems such as zebrafishes, chicken embryos and adult mice, indicating the evolutionary pres- ervation of this gene regulation system in vivo.