Statistics of Exon Lengths in Fungi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Jpn. J. Protozool. 38(2) 171-183
Jpn. J. Protozool. Vol. 38, No. 2. (2005) 171 Review On the origin of mitochondria and Rickettsia-related eukaryotic endo- symbionts B. Franz Lang1*, Henner Brinkmann1, Liisa B. Koski1, Masahiro Fujishima2, Hans-Dieter Görtz3 and Gertraud Burger1 1Program in Evolutionary Biology, Canadian Institute for Advanced Research; Centre Robert Cedergren, Département de Biochimie, Université de Montréal, 2900 Boulevard Edouard- Montpetit, Montréal, Québec, H3T 1J4, Canada. 2Biological Institute, Faculty of Science, Yamaguchi University, Yamaguchi 753-8512, Japan. 3Abteilung Zoologie, Biologisches Institut, Universität Stuttgart, Pfaffenwaldring 57, 70550 Stuttgart, Germany. SUMMARY sence of genes for oxidative phosphorylation, the TCA cycle, and many other metabolic pathways, Resent insights into the origin and early evo- but the presence of several pathogenesis-related lution of mitochondria come from two approaches: genes and a high number of bacterial IS elements. the investigation of mtDNAs from minimally de- Phylogenetic analyses with multiple protein se- rived (primitive) mitochondriate eukaryotes, in quences place H. obtusa basally to the Rickettsia- particular jakobid flagellates, and of genomes from Ehrlichia-Wolbachia assemblage of bacterial intracellular α-proteobacterial symbionts. Of par- pathogens. This leads us to postulate that H. ob- ticular interest in this context is Holospora obtusa, tusa is the closest bacterial relative of mitochon- an intracellular bacterial endosymbiont that resides dria known to date. and replicates in the somatic nucleus of its eu- karyotic host, the ciliate Paramecium caudatum. Currently we have sequenced close to 50% of the INTRODUCTION ~ 1.7 Mbp H. obtusa genome, revealing the ab- One of the major advancements in under- standing eukaryotic evolution was the discovery that mitochondria evolved from an endosymbiotic α-Proteobacterium, and that mitochondrial DNA *Corresponding author (mtDNA) is a relict bacterial genome. -

Sex Is a Ubiquitous, Ancient, and Inherent Attribute of Eukaryotic Life
PAPER Sex is a ubiquitous, ancient, and inherent attribute of COLLOQUIUM eukaryotic life Dave Speijera,1, Julius Lukešb,c, and Marek Eliášd,1 aDepartment of Medical Biochemistry, Academic Medical Center, University of Amsterdam, 1105 AZ, Amsterdam, The Netherlands; bInstitute of Parasitology, Biology Centre, Czech Academy of Sciences, and Faculty of Sciences, University of South Bohemia, 370 05 Ceské Budejovice, Czech Republic; cCanadian Institute for Advanced Research, Toronto, ON, Canada M5G 1Z8; and dDepartment of Biology and Ecology, University of Ostrava, 710 00 Ostrava, Czech Republic Edited by John C. Avise, University of California, Irvine, CA, and approved April 8, 2015 (received for review February 14, 2015) Sexual reproduction and clonality in eukaryotes are mostly Sex in Eukaryotic Microorganisms: More Voyeurs Needed seen as exclusive, the latter being rather exceptional. This view Whereas absence of sex is considered as something scandalous for might be biased by focusing almost exclusively on metazoans. a zoologist, scientists studying protists, which represent the ma- We analyze and discuss reproduction in the context of extant jority of extant eukaryotic diversity (2), are much more ready to eukaryotic diversity, paying special attention to protists. We accept that a particular eukaryotic group has not shown any evi- present results of phylogenetically extended searches for ho- dence of sexual processes. Although sex is very well documented mologs of two proteins functioning in cell and nuclear fusion, in many protist groups, and members of some taxa, such as ciliates respectively (HAP2 and GEX1), providing indirect evidence for (Alveolata), diatoms (Stramenopiles), or green algae (Chlor- these processes in several eukaryotic lineages where sex has oplastida), even serve as models to study various aspects of sex- – not been observed yet. -

BMC Research Notes Biomed Central
BMC Research Notes BioMed Central Short Report Open Access A split and rearranged nuclear gene encoding the iron-sulfur subunit of mitochondrial succinate dehydrogenase in Euglenozoa Ryan MR Gawryluk and Michael W Gray* Address: Department of Biochemistry and Molecular Biology, Dalhousie University, Halifax, Nova Scotia B3H 1X5, Canada Email: Ryan MR Gawryluk - [email protected]; Michael W Gray* - [email protected] * Corresponding author Published: 3 February 2009 Received: 18 December 2008 Accepted: 3 February 2009 BMC Research Notes 2009, 2:16 doi:10.1186/1756-0500-2-16 This article is available from: http://www.biomedcentral.com/1756-0500/2/16 © 2009 Gray et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: Analyses based on phylogenetic and ultrastructural data have suggested that euglenids (such as Euglena gracilis), trypanosomatids and diplonemids are members of a monophyletic lineage termed Euglenozoa. However, many uncertainties are associated with phylogenetic reconstructions for ancient and rapidly evolving groups; thus, rare genomic characters become increasingly important in reinforcing inferred phylogenetic relationships. Findings: We discovered that the iron-sulfur subunit (SdhB) of mitochondrial succinate dehydrogenase is encoded by a split and rearranged nuclear gene in Euglena gracilis and trypanosomatids, an example of a rare genomic character. The two subgenic modules are transcribed independently and the resulting mRNAs appear to be independently translated, with the two protein products imported into mitochondria, based on the presence of predicted mitochondrial targeting peptides. -

Role of Lipids and Fatty Acids in Stress Tolerance in Cyanobacteria
Acta Protozool. (2002) 41: 297 - 308 Review Article Role of Lipids and Fatty Acids in Stress Tolerance in Cyanobacteria Suresh C. SINGH, Rajeshwar P. SINHA and Donat-P. HÄDER Institut für Botanik und Pharmazeutische Biologie, Friedrich-Alexander-Universität, Erlangen, Germany Summary. Lipids are the most effective source of storage energy, function as insulators of delicate internal organs and hormones and play an important role as the structural constituents of most of the cellular membranes. They also have a vital role in tolerance to several physiological stressors in a variety of organisms including cyanobacteria. The mechanism of desiccation tolerance relies on phospholipid bilayers which are stabilized during water stress by sugars, especially by trehalose. Unsaturation of fatty acids also counteracts water or salt stress. Hydrogen atoms adjacent to olefinic bonds are susceptible to oxidative attack. Lipids are rich in these bonds and are a primary target for oxidative reactions. Lipid oxidation is problematic as enzymes do not control many oxidative chemical reactions and some of the products of the attack are highly reactive species that modify proteins and DNA. This review deals with the role of lipids and fatty acids in stress tolerance in cyanobacteria. Key words: cyanobacteria, desiccation, fatty acids, lipids, salinity, temperature stress. INTRODUCTION The cyanobacteria such as Spirulina and Nostoc have been used as a source of protein and vitamin for Cyanobacteria are gram-negative photoautotrophic humans and animals (Ciferri 1983, Kay 1991, Gao 1998, prokaryotes having ´higher plant-type‘ oxygenic photo- Takenaka et al. 1998). Spirulina has an unusually high synthesis (Stewart 1980, Sinha and Häder 1996a). -

Origin and Diversification of Eukaryotes
MI66CH20-Katz ARI 21 June 2012 17:22 V I E E W R S Review in Advance first posted online on July 9, 2012. (Changes may still occur before final publication E online and in print.) I N C N A D V A Origin and Diversification of Eukaryotes Laura A. Katz Department of Biological Sciences, Smith College, Northampton, Massachusetts 01063; email: [email protected] Program in Organismic and Evolutionary Biology, University of Massachusetts, Amherst, Massachusetts 01003 Annu. Rev. Microbiol. 2012. 66:411–27 Keywords The Annual Review of Microbiology is online at eukaryotic diversity, protists, tree of life, nucleus, cytoskeleton, micro.annualreviews.org mitochondria This article’s doi: by SMITH COLLEGE on 08/12/12. For personal use only. 10.1146/annurev-micro-090110-102808 Abstract Copyright c 2012 by Annual Reviews. The bulk of the diversity of eukaryotic life is microbial. Although the larger All rights reserved Annu. Rev. Microbiol. 2012.66. Downloaded from www.annualreviews.org eukaryotes—namely plants, animals, and fungi—dominate our visual land- 0066-4227/12/1013-0411$20.00 scapes, microbial lineages compose the greater part of both genetic diversity and biomass, and contain many evolutionary innovations. Our understand- ing of the origin and diversification of eukaryotes has improved substan- tially with analyses of molecular data from diverse lineages. These data have provided insight into the nature of the genome of the last eukaryotic com- mon ancestor (LECA). Yet, the origin of key eukaryotic features, namely the nucleus and cytoskeleton, remains poorly understood. In contrast, the past decades have seen considerable refinement in hypotheses on the major branching events in the evolution of eukaryotic diversity. -

Organellar Genome Evolution in Red Algal Parasites: Differences in Adelpho- and Alloparasites
University of Rhode Island DigitalCommons@URI Open Access Dissertations 2017 Organellar Genome Evolution in Red Algal Parasites: Differences in Adelpho- and Alloparasites Eric Salomaki University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Salomaki, Eric, "Organellar Genome Evolution in Red Algal Parasites: Differences in Adelpho- and Alloparasites" (2017). Open Access Dissertations. Paper 614. https://digitalcommons.uri.edu/oa_diss/614 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. ORGANELLAR GENOME EVOLUTION IN RED ALGAL PARASITES: DIFFERENCES IN ADELPHO- AND ALLOPARASITES BY ERIC SALOMAKI A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN BIOLOGICAL SCIENCES UNIVERSITY OF RHODE ISLAND 2017 DOCTOR OF PHILOSOPHY DISSERTATION OF ERIC SALOMAKI APPROVED: Dissertation Committee: Major Professor Christopher E. Lane Jason Kolbe Tatiana Rynearson Nasser H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2017 ABSTRACT Parasitism is a common life strategy throughout the eukaryotic tree of life. Many devastating human pathogens, including the causative agents of malaria and toxoplasmosis, have evolved from a photosynthetic ancestor. However, how an organism transitions from a photosynthetic to a parasitic life history strategy remains mostly unknown. Parasites have independently evolved dozens of times throughout the Florideophyceae (Rhodophyta), and often infect close relatives. This framework enables direct comparisons between autotrophs and parasites to investigate the early stages of parasite evolution. -

10531 2007 9250 Article-Web 1..16
Biodivers Conserv (2008) 17:261–276 DOI 10.1007/s10531-007-9250-y ORIGINAL PAPER “Missing” protists: a molecular prospective Slava Epstein · PuriWcación López-García Received: 13 March 2007 / Accepted: 30 May 2007 / Published online: 24 October 2007 © Springer Science+Business Media B.V. 2007 Abstract Molecular ecology methods based on 18S rRNA ampliWcation and sequencing have revealed an astounding diversity of microbial eukaryotes in every environment sam- pled so far. This is certainly true of new species and genera, as essentially every new survey discovers a wealth of novel diversity at this level. This is almost certain for taxa that are higher in taxonomic hierarchy, as many molecular surveys reported novel clades within established protistan phyla, with some of these clades repeatedly conWrmed by subsequent studies. It may also be that the molecular approaches discovered several lineages of the highest taxonomic order, but this claim has not been vigorously veriWed as yet. Overall, the Weld of protistan diversity remains in its infancy. The true scale of this diversity is unknown, and so are the distribution of this diversity, its patterns, spatial and temporal dynamics, and ecological role. The sampled diversity appears to be just the tip of the ice- berg, and this oVers outstanding opportunities for microbial discovery for the purposes of both basic and applied research. Keywords 18S rRNA · Cryptic species · Eukaryotic phylogeny · Molecular ecology · Species richness Introduction Molecular analyses based on the rRNA approach have become an aVordable and practical tool that is used routinely to study microbial diversity in environmental samples. This type Special Issue: Protist diversity and geographic distribution. -
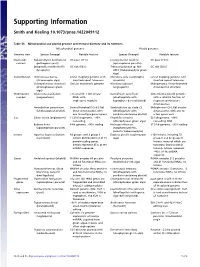
Supporting Information
Supporting Information Smith and Keeling 10.1073/pnas.1422049112 Table S1. Mitochondrial and plastid genome architectural diversity and its extremes Mitochondrial genomes Plastid genomes Genomic trait Species (lineage) Notable feature Species (lineage) Notable feature Nucleotide Nakaseomyces bacillisporus GC poor (11%) Leucocytozoon caulleryi GC poor (15%) content (pathogenic yeast) (apicomplexan parasite) Selaginella moellendorffii GC rich (68%) Trebouxiophyceae sp. MX- GC rich (58%) (lycophyte) AZ01 (trebouxiophyte green alga) Conformation Ochromonas danica Linear-mapping genome with Chromera velia (autotrophic Linear-mapping genome with (stramenopile alga) inverted-repeat telomeres alveolate) inverted repeat telomeres Chlamydomonas moewusii Circular monomeric genome Nicotiana tabacum Multigenomic, linear-branched (chlorophycean green (angiosperm) chromosomal structure alga) Chromosome Diplonema papillatum >75 small (6–7 kb) circular Karlodinium veneficum Conventional plastid genome number (euglenozoan) DNAs with (dinoflagellate with with a satellite fraction of single-gene modules haptophyte-derived plastid) fragmented miniature chromosomes Amoebidium parasiticum Several hundred (0.3–8.3 kb) Symbiodinium sp. clade C3 Multiple mini (2–3 kb) circular (ichthyosporean protist) linear chromosomes with (dinoflagellate with chromosomes with one to one to multiple genes apiece peridinin-containing plastid) a few genes each Size Silene conica (angiosperm) 11,318-kb genome, ∼99% Floydiella terrestris 521 kb genome, ∼80% noncoding (chlorophycean green alga) noncoding DNA Babesia bovis 6-kb genome, >90% coding Helicosporidium sp. 37.5 kb genome, ∼95% coding (apicomplexan parasite) (nonphotosynthetic, parasitic trebouxiophyte) Introns Agaricus bisporus (button 43 group I and 3 group II Euglena gracilis (euglenozoan >150 introns, including 72 mushroom) introns distributed in 8 of 15 alga) group II and 46 group III protein-coding genes. introns, many of which are Introns represent 45% of the twintrons (introns within mtDNA sequence introns). -

Systema Naturae. the Classification of Living Organisms
Systema Naturae. The classification of living organisms. c Alexey B. Shipunov v. 5.601 (June 26, 2007) Preface Most of researches agree that kingdom-level classification of living things needs the special rules and principles. Two approaches are possible: (a) tree- based, Hennigian approach will look for main dichotomies inside so-called “Tree of Life”; and (b) space-based, Linnaean approach will look for the key differences inside “Natural System” multidimensional “cloud”. Despite of clear advantages of tree-like approach (easy to develop rules and algorithms; trees are self-explaining), in many cases the space-based approach is still prefer- able, because it let us to summarize any kinds of taxonomically related da- ta and to compare different classifications quite easily. This approach also lead us to four-kingdom classification, but with different groups: Monera, Protista, Vegetabilia and Animalia, which represent different steps of in- creased complexity of living things, from simple prokaryotic cell to compound Nature Precedings : doi:10.1038/npre.2007.241.2 Posted 16 Aug 2007 eukaryotic cell and further to tissue/organ cell systems. The classification Only recent taxa. Viruses are not included. Abbreviations: incertae sedis (i.s.); pro parte (p.p.); sensu lato (s.l.); sedis mutabilis (sed.m.); sedis possi- bilis (sed.poss.); sensu stricto (s.str.); status mutabilis (stat.m.); quotes for “environmental” groups; asterisk for paraphyletic* taxa. 1 Regnum Monera Superphylum Archebacteria Phylum 1. Archebacteria Classis 1(1). Euryarcheota 1 2(2). Nanoarchaeota 3(3). Crenarchaeota 2 Superphylum Bacteria 3 Phylum 2. Firmicutes 4 Classis 1(4). Thermotogae sed.m. 2(5). -

Abstract Investigation of a Putative Mitochondrial Twin
ABSTRACT INVESTIGATION OF A PUTATIVE MITOCHONDRIAL TWIN ARGININE TRANSLOCATION PATHWAY IN ARABIDOPSIS THALIANA by Tasmeen Shiny Weerakoon The unique Twin Arginine Translocation (Tat) pathway which is composed of TatA, TatB and TatC, transports fully folded proteins without the use of ATP energy. It was thought to be lost from the mitochondrion, the powerhouse of the cell during post-endosymbiotic evolution. This study investigates the stress induced expression of a minimal, mitochondrial Tat pathway in Arabidopsis thaliana. We treated plants with salicylic acid (SA) stress which mimics the hypersensitive response and studied the putative mitoTat components over a 24-hour time period. qRT-PCR analysis indicated the upregulation of the mitochondrial gene mtTATC. Furthermore, immunoblot detection confirmed that the mtTATC protein is expressed at high SA concentrations. However, a minimal Tat system capable of translocation requires TatC as well as TatA (Tha4). Confocal microscopy data established the dual localization of nuclear encoded, chloroplast Tha4-GFP to the mitochondria. Together, our data illustrate the existence of a putative minimal mitoTat pathway at 4 hours post-treatment with high SA concentrations. Finally, we discuss the physiological significance of a SA-induced minimal mitoTat pathway in plant pathology. INVESTIGATION OF A PUTATIVE MITOCHONDRIAL TWIN ARGININE TRANSLOCATION PATHWAY IN ARABIDOPSIS THALIANA A Thesis Submitted to the Faculty of Miami University in partial fulfillment of the requirements for the degree of Master -

Inferring Ancestry
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1176 Inferring Ancestry Mitochondrial Origins and Other Deep Branches in the Eukaryote Tree of Life DING HE ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-554-9031-7 UPPSALA urn:nbn:se:uu:diva-231670 2014 Dissertation presented at Uppsala University to be publicly examined in Fries salen, Evolutionsbiologiskt centrum, Norbyvägen 18, 752 36, Uppsala, Friday, 24 October 2014 at 10:30 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Andrew Roger (Dalhousie University). Abstract He, D. 2014. Inferring Ancestry. Mitochondrial Origins and Other Deep Branches in the Eukaryote Tree of Life. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1176. 48 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-554-9031-7. There are ~12 supergroups of complex-celled organisms (eukaryotes), but relationships among them (including the root) remain elusive. For Paper I, I developed a dataset of 37 eukaryotic proteins of bacterial origin (euBac), representing the conservative protein core of the proto- mitochondrion. This gives a relatively short distance between ingroup (eukaryotes) and outgroup (mitochondrial progenitor), which is important for accurate rooting. The resulting phylogeny reconstructs three eukaryote megagroups and places one, Discoba (Excavata), as sister group to the other two (neozoa). This rejects the reigning “Unikont-Bikont” root and highlights the evolutionary importance of Excavata. For Paper II, I developed a 150-gene dataset to test relationships in supergroup SAR (Stramenopila, Alveolata, Rhizaria). Analyses of all 150-genes give different trees with different methods, but also reveal artifactual signal due to extremely long rhizarian branches and illegitimate sequences due to horizontal gene transfer (HGT) or contamination. -

AQPX-Cluster Aquaporins and Aquaglyceroporins Are
ARTICLE https://doi.org/10.1038/s42003-021-02472-9 OPEN AQPX-cluster aquaporins and aquaglyceroporins are asymmetrically distributed in trypanosomes ✉ ✉ Fiorella Carla Tesan 1,2, Ramiro Lorenzo 3, Karina Alleva 1,2,4 & Ana Romina Fox 3,4 Major Intrinsic Proteins (MIPs) are membrane channels that permeate water and other small solutes. Some trypanosomatid MIPs mediate the uptake of antiparasitic compounds, placing them as potential drug targets. However, a thorough study of the diversity of these channels is still missing. Here we place trypanosomatid channels in the sequence-function space of the large MIP superfamily through a sequence similarity network. This analysis exposes that trypanosomatid aquaporins integrate a distant cluster from the currently defined MIP 1234567890():,; families, here named aquaporin X (AQPX). Our phylogenetic analyses reveal that trypano- somatid MIPs distribute exclusively between aquaglyceroporin (GLP) and AQPX, being the AQPX family expanded in the Metakinetoplastina common ancestor before the origin of the parasitic order Trypanosomatida. Synteny analysis shows how African trypanosomes spe- cifically lost AQPXs, whereas American trypanosomes specifically lost GLPs. AQPXs diverge from already described MIPs on crucial residues. Together, our results expose the diversity of trypanosomatid MIPs and will aid further functional, structural, and physiological research needed to face the potentiality of the AQPXs as gateways for trypanocidal drugs. 1 Universidad de Buenos Aires, Facultad de Farmacia y Bioquímica, Departamento de Fisicomatemática, Cátedra de Física, Buenos Aires, Argentina. 2 CONICET-Universidad de Buenos Aires, Instituto de Química y Fisicoquímica Biológicas (IQUIFIB), Buenos Aires, Argentina. 3 Laboratorio de Farmacología, Centro de Investigación Veterinaria de Tandil (CIVETAN), (CONICET-CICPBA-UNCPBA) Facultad de Ciencias Veterinarias, Universidad Nacional del Centro ✉ de la Provincia de Buenos Aires, Tandil, Argentina.