RNA Splice Sites Classification Using Convolutional Neural Network Models
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

News & Views Research
NEWS & VIEWS RESEARCH may exhibit some tissue specificity in humans. Upstream intron Downstream intron a Sibley et al. found that genes with long introns sequence sequence tend to be expressed in the human nervous Precursor RNA system, and they identified recursively spliced 3' Splice 5' Splice RNAs expressed in the human brain6. Duff site site First step et al. detected some selectivity for recursive splicing in the brain in a screen of 20 human tissues (including fetal brain and adult cerebel- Second step lum), but this may partly reflect the difficulty of detecting recursively spliced RNAs in tis- Mature mRNA sues that express such RNAs at low levels. It will be important to determine whether this b RS exon specificity, if real, results from the tendency of recursively spliced genes to be expressed in the brain, or whether cells in the nervous system have factors that promote recursive Competing 5' splice sites First step splicing. Many genes that have long introns, including those that undergo recursive splicing, are linked to neurological diseases and to Second step 9–11 Second step autism . Whether these conditions are sometimes triggered by errors in the multi- step recursive RNA-splicing process will be an exciting avenue for future studies. ■ NMD Heidi Cook-Andersen and Miles F. Wilkinson are in the Department of Reproductive Medicine, University of Figure 1 | Mechanisms of recursive splicing. a, In recursive splicing, long intron sequences of precursor California, San Diego, La Jolla, California, RNA are removed in a stepwise process mediated by juxtaposed internal 3ʹ and 5ʹ splice sites. In the first step, 92093, USA. -

(Bsc Zoology and Microbiology) Concept of Introns and Exons
Unit-5 Molecular Biology (BSc Zoology and Microbiology) Concept of introns and exons Most of the portion of a gene in higher eukaryotes consists of noncoding DNA that interrupts the relatively short segments of coding DNA. The coding sequences are called exons. The noncoding sequences are called introns. Intron: An intron is a portion of a gene that does not code for amino acids An intron is any nucleotide sequence within a gene which is represented in the primary transcript of the gene, but not present in the final processed form. In other words, Introns are noncoding regions of an RNA transcript which are eliminated by splicing before translation. Sequences that are joined together in the final mature RNA after RNA splicing are exons. Introns are very large chunks of RNA within a messenger RNA molecule that interfere with the code of the exons. And these introns get removed from the RNA molecule to leave a string of exons attached to each other so that the appropriate amino acids can be encoded for. Introns are rare in genes of prokaryotes. #Look carefully at the diagram above, we have already discussed about the modification and processing of eukaryotic RNA. In which 5’ guanine cap and 3’poly A tail is added. So at that time, noncoding regions i.e. introns are removed. We hv done ths already. Ok Exon: The coding sequences are called Exon. An exon is the portion of a gene that codes for amino acids. In the cells of plants and animals, most gene sequences are broken up by one or more DNA sequences called introns. -

Current Perspectives in Intronic Micro Rnas (Mirnas)
Journal of Biomedical Science (2006) 13:5–15 5 DOI 10.1007/s11373-005-9036-8 Current perspectives in intronic micro RNAs (miRNAs) Shao-Yao Ying & Shi-Lung Lin Department of Cell & Neurobiology, Keck School of Medicine, BMT-403, University of Southern California, 1333 San Pablo Street, Los Angeles, CA, 90033, USA Received 27 May 2005; accepted 14 September 2005 Ó 2005 National Science Council, Taipei Key words: fine-tuning of gene function, functional/structural genomics, gene expression, genetic regula- tion, intronic microRNA, miRNA biogenesis, miRNA, post-translational modification, regulatory gene Summary MicroRNAs (miRNAs), small single-stranded regulatory RNAs capable of interfering with intracellular messenger RNAs (mRNAs) that contain either complete or partial complementarity, are useful for the design of new therapies against cancer polymorphism and viral mutation. Numerous miRNAs have been reported to induce RNA interference (RNAi), a post-transcriptional gene silencing mechanism. Intronic miRNAs, derived from introns by RNA splicing and Dicer processing, can interfere with intracellular mRNAs to silence that gene expression. The intronic miRNAs differ uniquely from previously described intergenic miRNAs in the requirement of type II RNA polymerases (Pol-II) and spliceosomal components for its biogenesis. Several kinds of intronic miRNAs have been identified in Caenorhabditis elegans, mouse and human cells; however, neither their function nor application has been reported. To this day, the computer searching program for miRNA seldom include the intronic portion of protein-coding RNAs. The functional significance of artificially generated intronic miRNAs has been successfully ascertained in several biological systems such as zebrafishes, chicken embryos and adult mice, indicating the evolutionary pres- ervation of this gene regulation system in vivo. -

The CTD Role in Cotranscriptional RNA Processing and Surveillance
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector FEBS Letters 582 (2008) 1971–1976 Minireview The CTD role in cotranscriptional RNA processing and surveillance Se´rgio F. de Almeida, Maria Carmo-Fonseca* Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal Received 4 April 2008; revised 13 April 2008; accepted 14 April 2008 Available online 22 April 2008 Edited by Ulrike Kutay proteins specific to each snRNP. The selection of specific splice Abstract In higher eukaryotes, the production of mature mes- senger RNA that exits the nucleus to be translated into protein sites (ss) on a particular pre-mRNA substrate relies on an intri- requires precise and extensive processing of the nascent tran- cate interplay involving the cooperative binding of trans-acting script. The processing steps include 50-end capping, splicing, splicing proteins to cis-acting sequence elements in the pre- and 30-end formation. Pre-mRNA processing is coupled to tran- mRNA. In mammals, an intron is defined by four short and scription by mechanisms that are not well understood but involve poorly conserved consensus sequences: the exon–intron junc- the carboxyl-terminal domain (CTD) of the largest subunit of tions (50ss and 30ss); the branch point sequence; and the poly- RNA polymerase II. This review focuses on recent findings that pyrimidine tract (Table 1). These sequences are recognized by provide novel insight into the role of the CTD in promoting RNA base pairing with the spliceosomal snRNAs (Fig. 1). In addi- processing and surveillance. tion, both exons and introns contain weak binding sites for a Ó 2008 Federation of European Biochemical Societies. -
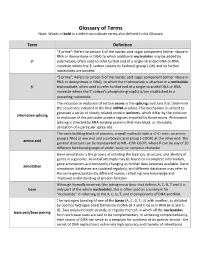
Glossary of Terms Note: Words in Bold in a Definition Indicate Terms Also Defined in This Glossary
Glossary of Terms Note: Words in bold in a definition indicate terms also defined in this Glossary Term Definition “3 prime”; Refers to carbon 3 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA) to which additional nucleotides may be added by 3' polymerase, often used to refer to that end of a single-stranded DNA or RNA molecule where the 3' carbon retains its hydroxyl group (-OH) and no further nucleotides are bonded. “5 prime”; Refers to carbon 5 of the nucleic acid sugar component (either ribose in RNA or deoxyribose in DNA), to which the triphosphate is attached in a nucleotide 5' triphosphate, often used to refer to that end of a single-stranded DNA or RNA molecule where the 5' carbon's phosphate group(s) is/are unattached to a preceding nucleotide. The inclusion or exclusion of certain exons in the splicing reactions that determine the sequences included in the final mRNA product. This mechanism is utilized to generate a series of closely related protein isoforms, which differ by the inclusion alternative splicing or exclusion of the particular protein regions encoded by those exons. Alternative splicing is directed by RNA-binding proteins that may block, or stimulate, utilization of a particular splice site. The basic building block of proteins, a small molecule with a -C-C- core, an amine group (-NH2) at one end and a carboxylic acid group (-COOH) at the other end. The amino acid general structure can be represented as NH2-CHR-COOH, where R can be any of 20 different functional groups of acidic, basic, or nonpolar character. -

Protein-Synthesis.Pdf
Protein Synthesis Contents Introduction ........................................................................................................................ 1 DNA Transcription ............................................................................................................... 1 The Primary Transcript ........................................................................................................ 4 Translation .......................................................................................................................... 7 Protein Structure............................................................................................................... 11 References ........................................................................................................................ 11 Resources .......................................................................................................................... 11 Introduction The genetic message carried on the DNA molecule is a code. This code is ultimately translated into a sequence of amino acids that, when complete, becomes a protein. Proteins carry out the “business” of the cell. Some proteins are used as structural components of cells, some are used to transport other molecules, still others are charged with directing chemical reactions. The latter class of proteins is the enzymes. Regardless of the role played by a protein in the cell one aspect is the same, they are all encoded in the base sequences of DNA. The path from DNA sequence to protein -

The Chloroplast Trans-Splicing RNA–Protein Supercomplex from the Green Alga Chlamydomonas Reinhardtii
cells Review The Chloroplast Trans-Splicing RNA–Protein Supercomplex from the Green Alga Chlamydomonas reinhardtii Ulrich Kück * and Olga Schmitt Allgemeine und Molekulare Botanik, Faculty for Biology and Biotechnology, Ruhr-Universität Bochum, Universitätsstraße 150, 44780 Bochum, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-234-32-28951 Abstract: In eukaryotes, RNA trans-splicing is a significant RNA modification process for the end-to- end ligation of exons from separately transcribed primary transcripts to generate mature mRNA. So far, three different categories of RNA trans-splicing have been found in organisms within a diverse range. Here, we review trans-splicing of discontinuous group II introns, which occurs in chloroplasts and mitochondria of lower eukaryotes and plants. We discuss the origin of intronic sequences and the evolutionary relationship between chloroplast ribonucleoprotein complexes and the nuclear spliceosome. Finally, we focus on the ribonucleoprotein supercomplex involved in trans-splicing of chloroplast group II introns from the green alga Chlamydomonas reinhardtii. This complex has been well characterized genetically and biochemically, resulting in a detailed picture of the chloroplast ribonucleoprotein supercomplex. This information contributes substantially to our understanding of the function of RNA-processing machineries and might provide a blueprint for other splicing complexes involved in trans- as well as cis-splicing of organellar intron RNAs. Keywords: group II intron; trans-splicing; ribonucleoprotein complex; chloroplast; Chlamydomonas reinhardtii Citation: Kück, U.; Schmitt, O. The Chloroplast Trans-Splicing RNA–Protein Supercomplex from the Green Alga Chlamydomonas reinhardtii. 1. Introduction Cells 2021, 10, 290. https://doi.org/ One of the unexpected and outstanding discoveries in 20th century biology was the 10.3390/cells10020290 identification of discontinuous eukaryotic genes [1,2]. -

Alternatively Spliced Genes
1 Alternatively Spliced Genes Jane Y. Wu1,2,LiyaYuan1 and Necat Havlioglu1 1Washington University School of Medicine, St. Louis, MO, USA 2John F. Kennedy Center for Research on Human Development, Vanderbilt University Medical Center, Nashville, TN, USA 1 Pre-mRNA Splicing and Splicing Machinery 3 1.1 Splicing Machinery: Spliceosome 3 1.2 Splicing Signals 4 1.3 Spliceosomal UsnRNP Biogenesis 12 1.4 Spliceosome Assembly 13 1.5 Biochemical Mechanisms of pre-mRNA Splicing 17 2 Alternative pre-mRNA Splicing 17 2.1 Alternative Splicing and its Role in Regulating Gene Activities and Generating Genetic Diversity 17 2.1.1 Different Patterns of Alternative Splicing 17 2.1.2 Alternative Splicing and Genetic Diversity 18 2.2 Mechanisms Underlying Alternative Splicing Regulation 19 2.2.1 Splicing Signals and Splicing Regulatory Elements 20 2.2.2 Trans-acting Splicing Regulators 23 2.3 Tissue-specific and Developmentally Regulated Alternative Splicing 26 2.4 Regulation of Alternative Splicing in Response to Extracellular Stimuli 27 3 Pre-mRNA Splicing and Human Diseases 28 3.1 Splicing Defects in Human Diseases 28 3.2 Molecular Mechanisms Underlying Splicing Defects Associated with Disease 33 4 Perspectives on Diagnosis and Treatment of Diseases Caused by pre-mRNA Splicing Defects 36 4.1 Diagnosis of Human Diseases Caused by Splicing Defects 36 2 Alternatively Spliced Genes 4.2 Potential Therapeutic Approaches 37 4.2.1 Oligonucleotide-based Approaches: Antisense, RNAi, and Chimeric Molecules 37 4.2.2 Ribozymes 37 4.2.3 SMaRT 38 4.2.4 Chemical Compounds 38 5 Concluding Remarks 38 Acknowledgment 39 Bibliography 39 Books and Reviews 39 Keywords Pre-mRNA Nascent transcripts that are precursors of mature messenger RNAs. -

Spliceosomal Pre-Mrna Splicing Methods and Protocols M ETHODS in MOLECULAR BIOLOGY
Methods in Molecular Biology 1126 Klemens J. Hertel Editor Spliceosomal Pre-mRNA Splicing Methods and Protocols M ETHODS IN MOLECULAR BIOLOGY Series Editor John M. Walker School of Life Sciences University of Hertfordshire Hat fi eld, Hertfordshire, AL10 9AB, UK For further volumes: http://www.springer.com/series/7651 Spliceosomal Pre-mRNA Splicing Methods and Protocols Edited by Klemens J. Hertel Department of Microbiology & Molecular Genetics, University of California, Irvine, CA, USA Editor Klemens J. Hertel Department of Microbiology & Molecular Genetics University of California Irvine , CA , USA ISSN 1064-3745 ISSN 1940-6029 (electronic) ISBN 978-1-62703-979-6 ISBN 978-1-62703-980-2 (eBook) DOI 10.1007/978-1-62703-980-2 Springer New York Heidelberg Dordrecht London Library of Congress Control Number: 2014931088 © Springer Science+Business Media, LLC 2014 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifi cally the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfi lms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifi cally for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. Duplication of this publication or parts thereof is permitted only under the provisions of the Copyright Law of the Publisher’s location, in its current version, and permission for use must always be obtained from Springer. -

Post-Transcriptional Modification by / Hanaa Shawky Ragheb Supervised by Prof.Dr / Rasha El Tahan Course / Molecular Biology
Post-transcriptional modification By / Hanaa Shawky Ragheb Supervised by prof.dr / Rasha El Tahan Course / Molecular Biology Molecular biology: is a branch of science concerning biological activity at the molecular level. The field of molecular biology overlaps with biology and chemistry and in particular, genetics and biochemistry. A key area of molecular biology concerns understanding how various cellular systems interact in terms of the way DNA, RNA and protein synthesis function. Molecular biology looks at the molecular mechanisms behind processes such as replication, transcription, translation and cell function. One way to describe the basis of molecular biology is to say it concerns understanding how genes are transcribed into RNA and how RNA is then translated into protein. However, this simplified picture is currently be reconsidered and revised due to new discoveries concerning the roles of RNA. Central Dogma (Gene Expression): The central dogma of molecular biology explains that the information flow for genes is from the DNA genetic code to an intermediate RNA copy and then to the proteins synthesized from the code. The key ideas underlying the dogma were first proposed by British molecular biologist Francis Crick in 1958. Replication : DNA replication The formation of new and, hopefully, identical copies of complete genomes. DNA replication occurs every time a cell divides to form two daughter cells. Under the influence of enzymes, DNA unwinds and the two strands separate over short lengths to form numerous replication forks, each of which is called a replicon. The separated strands are temporarily sealed with protein to prevent re-attachment. A short RNA sequence called a primer is formed for each strand at the fork. -

RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays
molecules Review RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays Zhichao Tang, Junxing Zhao , Zach J. Pearson, Zarko V. Boskovic and Jingxin Wang * Department of Medicinal Chemistry, University of Kansas, Lawrence, KS 66047, USA; [email protected] (Z.T.); [email protected] (J.Z.); [email protected] (Z.J.P.); [email protected] (Z.V.B.) * Correspondence: [email protected] Abstract: RNA splicing is an essential step in producing mature messenger RNA (mRNA) and other RNA species. Harnessing RNA splicing modifiers as a new pharmacological modality is promising for the treatment of diseases caused by aberrant splicing. This drug modality can be used for infectious diseases by disrupting the splicing of essential pathogenic genes. Several antisense oligonucleotide splicing modifiers were approved by the U.S. Food and Drug Administration (FDA) for the treatment of spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD). Recently, a small-molecule splicing modifier, risdiplam, was also approved for the treatment of SMA, highlighting small molecules as important warheads in the arsenal for regulating RNA splicing. The cellular targets of these approved drugs are all mRNA precursors (pre-mRNAs) in human cells. The development of novel RNA-targeting splicing modifiers can not only expand the scope of drug targets to include many previously considered “undruggable” genes but also enrich the chemical-genetic toolbox for basic biomedical research. In this review, we summarized known Citation: Tang, Z.; Zhao, J.; Pearson, splicing modifiers, screening methods for novel splicing modifiers, and the chemical space occupied Z.J.; Boskovic, Z.V.; Wang, J. by the small-molecule splicing modifiers. -

The Role of 4E-T in Translational Regulation and Mrna Decay
The role of 4E-T in translational regulation and mRNA decay Dissertation der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) vorgelegt von Felix Alexander Paul Räsch aus Hermeskeil Tübingen 2020 Gedruckt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Eberhard Karls Universität Tübingen. Tag der mündlichen Qualifikation: 05.08.2020 Dekan: Prof. Dr. Wolfgang Rosenstiel 1. Berichterstatter: Prof. Dr. Ralf-Peter Jansen 2. Berichterstatter: Prof. Dr. Patrick Müller This thesis describes work conducted in the laboratory of Prof. Dr. Elisa Izaurralde in the Department of Biochemistry at the Max Planck Institute for Developmental Biology, Tübingen, Germany, from February 2015 to May 2020. The work was supervised by Prof. Dr. Ralf-Peter Jansen at the Eberhard Karls University Tübingen, Germany, and was supported by a fellowship from the International PhD program of the MPI for Developmental Biology. I declare that this thesis is the product of my own work. The parts that have been published or where other sources have been used were cited accordingly. Work carried out by my colleagues was also indicated accordingly. Acknowledgements Acknowledgements This thesis would not have been possible without all the support I received during the last years for which I will be forever grateful. I would like to express my deepest gratitude to Prof. Dr. Elisa Izaurralde for giving me the opportunity to work in her lab and giving me support and guidance that shaped my understanding of science more than anything. She was a great mentor who cared deeply about the growth of her PhD students and her devotion to science was truly inspiring for me.