Alternatively Spliced Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Protein Required for RNA Processing and Splicing in Neurospora
Proc. Nati. Acad. Sci. USA Vol. 89, pp. 1676-1680, March 1992 Genetics A protein required for RNA processing and splicing in Neurospora mitochondria is related to gene products involved in cell cycle protein phosphatase functions BEATRICE TURCQ*, KATHERINE F. DOBINSONt, NOBUFUSA SERIZAWAt, AND ALAN M. LAMBOWITZ§ Departments of Molecular Genetics and Biochemistry, and the Biotechnology Center, The Ohio State University, 484 West Twelfth Avenue, Columbus, OH 43210 Communicated by Thomas R. Cech, November 25, 1991 ABSTRACT The Neurospora crassa cyt4 mutants have specifically affect the splicing of group I introns and have pleiotropic defects in mitochondrial RNA splicing, 5' and 3' end relatively little effect on other mitochondrial RNA processing processing, and RNA turnover. Here, we show that the cyt-4 reactions (1, 4, 7). gene encodes a 120-kDa protein with significant similarity to By contrast, the cyt4 mutants have a complex phenotype the SSD1/SRK1 protein of Saceharomyces cerevisiae and the with defects in a number of different aspects of mitochondrial DIS3 protein of Sclhizosaccharomyces pombe, which have been RNA metabolism in addition to RNA splicing (9, 10). The five implicated in protein phosphatase functions that regulate cell cyt4 mutants are cold sensitive; they grow slowly at 250C and cycle and mitotic chromosome segregation. The CYT-4 protein more rapidly at 370C. Initial studies focusing on the mitochon- is present in mitochondria and is truncated or deficient in two drial large rRNA showed that the cyt4 mutants are defective cyt4 mutants. Assuming that the CYT-4 protein functions in a in both splicing and 3' end synthesis and that the splicing defect manner similar to the SSD1/SRK1 and DIS3 proteins, we infer is more pronounced at lower temperatures (9). -

Differential Analysis of Gene Expression and Alternative Splicing
i bioRxiv preprint doi: https://doi.org/10.1101/2020.08.23.234237; this version posted August 24, 2020. The copyright holder for this preprint i (which was not certified by peer review)“main” is the author/funder, — 2020/8/23 who has — granted 9:38 — bioRxiv page a1 license — #1 to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. i i EmpiReS: Differential Analysis of Gene Expression and Alternative Splicing Gergely Csaba∗, Evi Berchtold*,y, Armin Hadziahmetovic, Markus Gruber, Constantin Ammar and Ralf Zimmer Department of Informatics, Ludwig-Maximilians-Universitat¨ Munchen,¨ 80333, Munich, Germany ABSTRACT to translation. Different transcripts of the same gene are often expressed at the same time, possibly at different abundance, While absolute quantification is challenging in high- yielding a defined mixture of isoforms. Changes to this throughput measurements, changes of features composition are further referred to as differential alternative between conditions can often be determined with splicing (DAS). Various diseases can be linked to DAS high precision. Therefore, analysis of fold changes events (3). Most hallmarks of cancer like tumor progression is the standard method, but often, a doubly and immune escape (4, 5) can be attributed to changes in differential analysis of changes of changes is the transcript composition (6). DAS plays a prominent role required. Differential alternative splicing is an in various types of muscular dystrophy (MD) (7), splicing example of a doubly differential analysis, i.e. fold intervention by targeted exon removal is a therapeutic option changes between conditions for different isoforms in Duchenne MD (8). -

Analysis of Exonic Mutations Leading to Exon Skipping in Patients with Pyruvate Dehydrogenase E1␣ Deficiency
0031-3998/00/4806-0748 PEDIATRIC RESEARCH Vol. 48, No. 6, 2000 Copyright © 2000 International Pediatric Research Foundation, Inc. Printed in U.S.A. Analysis of Exonic Mutations Leading to Exon Skipping in Patients with Pyruvate Dehydrogenase E1␣ Deficiency ALESSANDRA KUPPER CARDOZO, LINDA DE MEIRLEIR, INGE LIEBAERS, AND WILLY LISSENS Center for Medical Genetics [A.K.C., L.D.M., I.L., W.L.] and Pediatric Neurology [L.D.M.], University Hospital, Vrije Universiteit Brussel, 1090 Brussels, Belgium. ABSTRACT The pyruvate dehydrogenase (PDH) complex is situated at a mutations that either reverted or disrupted the wild-type pre- key position in energy metabolism and is responsible for the dicted pre-mRNA secondary structure of exon 6, we were unable conversion of pyruvate to acetyl CoA. In the literature, two to establish a correlation between the aberrant splicing and unrelated patients with a PDH complex deficiency and splicing disruption of the predicted structure. The mutagenic experiments out of exon 6 of the PDH E1␣ gene have been described, described here and the silent mutation found in one of the although intronic/exonic boundaries on either side of exon 6 patients suggest the presence of an exonic splicing enhancer in were completely normal. Analysis of exon 6 in genomic DNA of the middle region of exon 6 of the PDH E1␣ gene. (Pediatr Res these patients revealed two exonic mutations, a silent and a 48: 748–753, 2000) missense mutation. Although not experimentally demonstrated, the authors in both publications suggested that the exonic muta- tions were responsible for the exon skipping. In this work, we Abbreviations were able to demonstrate, by performing splicing experiments, ESE, exonic splicing enhancer that the two exonic mutations described in the PDH E1␣ gene mfe, minimum free energy lead to aberrant splicing. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Transcription to RNA from Gene to Phenotype
11/8/11 Transcription to RNA From Gene to Phenotype DNA Gene 2 • The central dogma: molecule – DNA->RNA->protein Gene 1 – One Gene, One Enzyme Gene 3 • Beadle & Tatum expts. • Transcription DNA strand 3! 5! – Initiation (template) A C C A A A C C G A G T – Elongation – Termination TRANSCRIPTION U G G U U U G G C U C A • mRNA processing mRNA 5! 3! – Introns and exons Codon TRANSLATION • Other types of RNA 11/9/2011 Protein Trp Phe Gly Ser Amino acid In Prokaryotes transcription and translation occur simultaneously In Eukaryotes Nuclear – Transcription and envelope translation occur in separate compartments TRANSCRIPTION DNA DNA TRANSCRIPTION of the cell Pre-mRNA RNA PROCESSING mRNA Ribosome – RNA transcripts are mRNA TRANSLATION modified before becoming true mRNA Ribosome Polypeptide TRANSLATION Polypeptide Figure 17.3a Figure 17.3b One Gene -> One Enzyme “One Gene -> One Enzyme” EXPERIMENT Class I Class II Class III Wild type Mutants Mutants Mutants Minimal Normal bread-mold cells can medium synthesize arginine from precursors (MM) (control) in the minimal medium MM + Ornithine Mutant 2 could MM + grow if either Citrulline Precursor Ornithine Citrulline Arginine citruline or MM + arginine was Specific enzymes (arrows) Arginine is an essential Arginine amino acid, required for (control) supplied. catalyze each step growth Therefore it must lack the enzyme to make Citruline Precursor Ornithine Citrulline Arginine 1 11/8/11 From Gene to Phenotype RNA Polymerase Non-template strand of DNA DNA Gene 2 RNA nucleotides molecule Gene 1 Gene 3 T C C A A A T 3 C T U ! 3 end DNA strand 3! 5! ! G (template) A C C A A A C C G A G T T A U G G A 5 C A U C C A C TRANSCRIPTION ! A T A A G G T T U G G U U U G G C U C A mRNA 5! 3! Direction of transcription 5! Codon (“downstream) Template TRANSLATION strand of DNA Protein Trp Phe Gly Ser New RNA Amino acid Synthesis of an RNA Transcript DNA is copied to make messenger RNA Promoter Transcription unit 5! 3! 3! 5! RNA polymerase DNA This is the “non-template” Start point binds to a promoter RNA polymerase strand. -

Serine Arginine-Rich Protein-Dependent Suppression Of
Serine͞arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers El Che´ rif Ibrahim*, Thomas D. Schaal†, Klemens J. Hertel‡, Robin Reed*, and Tom Maniatis†§ *Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115; †Department of Molecular and Cellular Biology, Harvard University, 7 Divinity Avenue, Cambridge, MA 02138; and ‡Department of Microbiology and Molecular Genetics, University of California, Irvine, CA 92697-4025 Contributed by Tom Maniatis, January 28, 2005 The 5 and 3 splice sites within an intron can, in principle, be joined mechanisms by which exon skipping is prevented. Our data to those within any other intron during pre-mRNA splicing. How- reveal that splicing to the distal 3Ј splice site is suppressed by ever, exons are joined in a strict 5 to 3 linear order in constitu- proximal exonic sequences and that SR proteins are required for tively spliced pre-mRNAs. Thus, specific mechanisms must exist to this suppression. Thus, SR protein͞exonic enhancer complexes prevent the random joining of exons. Here we report that insertion not only function in exon and splice-site recognition but also play of exon sequences into an intron can inhibit splicing to the a role in ensuring that 5Ј and 3Ј splice sites within the same intron .downstream 3 splice site and that this inhibition is independent of are used, thus suppressing exon skipping intron size. The exon sequences required for splicing inhibition were found to be exonic enhancer elements, and their inhibitory Materials and Methods activity requires the binding of serine͞arginine-rich splicing fac- Construction of Plasmids. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

The Splicing Factor XAB2 Interacts with ERCC1-XPF and XPG for RNA-Loop Processing During Mammalian Development
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.20.211441; this version posted July 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The Splicing Factor XAB2 interacts with ERCC1-XPF and XPG for RNA-loop processing during mammalian development Evi Goulielmaki1*, Maria Tsekrekou1,2*, Nikos Batsiotos1,2, Mariana Ascensão-Ferreira3, Eleftheria Ledaki1, Kalliopi Stratigi1, Georgia Chatzinikolaou1, Pantelis Topalis1, Theodore Kosteas1, Janine Altmüller4, Jeroen A. Demmers5, Nuno L. Barbosa-Morais3, George A. Garinis1,2* 1. Institute of Molecular Biology and Biotechnology, Foundation for Research and Technology- Hellas, GR70013, Heraklion, Crete, Greece, 2. Department of Biology, University of Crete, Heraklion, Crete, Greece, 3. Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina da Universidade de Lisboa, Avenida Professor Egas Moniz, 1649-028 Lisboa, Portugal, 4. Cologne Center for Genomics (CCG), Institute for Genetics, University of Cologne, 50931, Cologne, Germany, 5. Proteomics Center, Netherlands Proteomics Center, and Department of Biochemistry, Erasmus University Medical Center, the Netherlands. Corresponding author: George A. Garinis ([email protected]) *: equally contributing authors bioRxiv preprint doi: https://doi.org/10.1101/2020.07.20.211441; this version posted July 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract RNA splicing, transcription and the DNA damage response are intriguingly linked in mammals but the underlying mechanisms remain poorly understood. Using an in vivo biotinylation tagging approach in mice, we show that the splicing factor XAB2 interacts with the core spliceosome and that it binds to spliceosomal U4 and U6 snRNAs and pre-mRNAs in developing livers. -
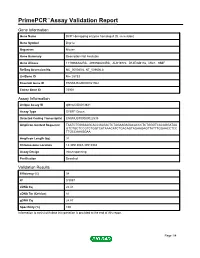
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name DCP1 decapping enzyme homolog A (S. cerevisiae) Gene Symbol Dcp1a Organism Mouse Gene Summary Description Not Available Gene Aliases 1110066A22Rik, 4930568L04Rik, AU019772, D14Ertd817e, Mitc1, SMIF RefSeq Accession No. NC_000080.6, NT_039606.8 UniGene ID Mm.28733 Ensembl Gene ID ENSMUSG00000021962 Entrez Gene ID 75901 Assay Information Unique Assay ID qMmuCID0013841 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000022535 Amplicon Context Sequence TAATCTGGGAAGCACCGAGACTCTAGAAGAGACACCCTCTGGGTCACAGGATAA GTCTGCTCCGTCTGGTCATAAACATCTGACAGTAGAAGAGTTATTTGGAACCTCC TTGCCAAAGGAA Amplicon Length (bp) 91 Chromosome Location 14:30513043-30518984 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 cDNA Cq 22.41 cDNA Tm (Celsius) 81 gDNA Cq 24.87 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Dcp1a, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Exon Amplification: a Strategy to Isolate Mammalian Genes Based on RNA Splicing (Gene Cloning/Polymerase Chain Reaction) ALAN J
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 4005-4009, May 1991 Genetics Exon amplification: A strategy to isolate mammalian genes based on RNA splicing (gene cloning/polymerase chain reaction) ALAN J. BUCKLER*, DAVID D. CHANG, SHARON L. GRAw, J. DAVID BROOK, DANIEL A. HABER, PHILLIP A. SHARP, AND DAVID E. HOUSMAN Center for Cancer Research, Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 Contributed by Phillip A. Sharp, January 25, 1991 ABSTRACT We have developed a method, exon amplifi- (7, 8). Thus, this method may be generally applicable for the cation, for fast and efficient isolation of coding sequences from selection of exon sequences from any gene. The method is complex mammalian genomic DNA. This method is based on also both rapid and easily adapted to large scale experiments. the selection of RNA sequences, exons, which are flanked by A series of cloned genomic DNA fragments can be screened functional 5' and 3' splice sites. Fragments of cloned genomic within 1-2 weeks. The sensitivity of this method is high. DNA are inserted into an intron, which is flanked by 5' and 3' Genomic DNA segments of 20 kilobases (kb) or more can be splice sites of the human immunodeficiency virus 1 tat gene successfully screened in a single transfection by using a set contained within the plasmid pSPL1. COS-7 cells are trans- of pooled subclones. This method thus allows the rapid fected with these constructs, and the resulting RNA transcripts identification of exons in mammalian genomic DNA and are processed in vivo. Splice sites of exons contained within the should facilitate the isolation of a wide spectrum of genes of inserted genomic fragment are paired with splice sites of the significance in physiology and development. -

Mrna Turnover Philip Mitchell* and David Tollervey†
320 mRNA turnover Philip Mitchell* and David Tollervey† Nuclear RNA-binding proteins can record pre-mRNA are cotransported to the cytoplasm with the mRNP. These processing events in the structure of messenger proteins may preserve a record of the nuclear history of the ribonucleoprotein particles (mRNPs). During initial rounds of pre-mRNA in the cytoplasmic mRNP structure. This infor- translation, the mature mRNP structure is established and is mation can strongly influence the cytoplasmic fate of the monitored by mRNA surveillance systems. Competition for the mRNA and is used by mRNA surveillance systems that act cap structure links translation and subsequent mRNA as a checkpoint of mRNP integrity, particularly in the identi- degradation, which may also involve multiple deadenylases. fication of premature translation termination codons (PTCs). Addresses Cotransport of nuclear mRNA-binding proteins with mRNA Wellcome Trust Centre for Cell Biology, ICMB, University of Edinburgh, from the nucleus to the cytoplasm (nucleocytoplasmic shut- Kings’ Buildings, Edinburgh EH9 3JR, UK tling) was first observed for the heterogeneous nuclear *e-mail: [email protected] ribonucleoprotein (hnRNP) proteins. Some hnRNP proteins †e-mail: [email protected] are stripped from the mRNA at export [1], but hnRNP A1, Current Opinion in Cell Biology 2001, 13:320–325 A2, E, I and K are all exported (see [2]). Although roles for 0955-0674/01/$ — see front matter these hnRNP proteins in transport and translation have been © 2001 Elsevier Science Ltd. All rights reserved. reported [3•,4•], their affects on mRNA stability have been little studied. More is known about hnRNP D/AUF1 and Abbreviations AREs AU-rich sequence elements another nuclear RNA-binding protein, HuR, which act CBC cap-binding complex antagonistically to modulate the stability of a range of DAN deadenylating nuclease mRNAs containing AU-rich sequence elements (AREs) DSEs downstream sequence elements (reviewed in [2]).