Mrna Turnover Philip Mitchell* and David Tollervey†
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Ribosome As a Regulator of Mrna Decay
www.nature.com/cr www.cell-research.com RESEARCH HIGHLIGHT Make or break: the ribosome as a regulator of mRNA decay Anthony J. Veltri1, Karole N. D’Orazio1 and Rachel Green 1 Cell Research (2020) 30:195–196; https://doi.org/10.1038/s41422-019-0271-3 Cells regulate α- and β-tubulin levels through a negative present. To address this, the authors mixed pre-formed feedback loop which degrades tubulin mRNA upon detection TTC5–tubulin RNCs containing crosslinker with lysates from of excess free tubulin protein. In a recent study in Science, Lin colchicine-treated or colchicine-untreated TTC5-knockout cells et al. discover a role for a novel factor, TTC5, in recognizing (either having or lacking abundant free tubulin, respectively). the N-terminal motif of tubulins as they emerge from the After irradiation, TTC5 only crosslinked to the RNC in lysates ribosome and in signaling co-translational mRNA decay. from cells that had previously been treated with colchicine; Cells use translation-coupled mRNA decay for both quality these data suggested to the authors that some other (unknown) control and general regulation of mRNA levels. A variety of known factor may prevent TTC5 from binding under conditions of low quality control pathways including Nonsense Mediated Decay free tubulin. (NMD), No-Go Decay (NGD), and Non-Stop Decay (NSD) specifi- What are likely possibilities for how such coupling between cally detect and degrade mRNAs encoding potentially toxic translation and mRNA decay might occur? One example to protein fragments or sequences which cause ribosomes to consider is that of mRNA surveillance where extensive studies in translate poorly or stall.1 More generally, canonical mRNA yeast have identified a large group of proteins that recognize degradation is broadly thought to be translation dependent, and resolve stalled RNCs found on problematic mRNAs and 1234567890();,: though the mechanisms that drive these events are not target those mRNAs for decay. -

Coupling of Spliceosome Complexity to Intron Diversity
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Coupling of spliceosome complexity to intron diversity Jade Sales-Lee1, Daniela S. Perry1, Bradley A. Bowser2, Jolene K. Diedrich3, Beiduo Rao1, Irene Beusch1, John R. Yates III3, Scott W. Roy4,6, and Hiten D. Madhani1,6,7 1Dept. of Biochemistry and Biophysics University of California – San Francisco San Francisco, CA 94158 2Dept. of Molecular and Cellular Biology University of California - Merced Merced, CA 95343 3Department of Molecular Medicine The Scripps Research Institute, La Jolla, CA 92037 4Dept. of Biology San Francisco State University San Francisco, CA 94132 5Chan-Zuckerberg Biohub San Francisco, CA 94158 6Corresponding authors: [email protected], [email protected] 7Lead Contact 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.03.19.436190; this version posted March 20, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. SUMMARY We determined that over 40 spliceosomal proteins are conserved between many fungal species and humans but were lost during the evolution of S. cerevisiae, an intron-poor yeast with unusually rigid splicing signals. We analyzed null mutations in a subset of these factors, most of which had not been investigated previously, in the intron-rich yeast Cryptococcus neoformans. -

Lin28b and Mir-142-3P Regulate Neuronal Differentiation by Modulating Staufen1 Expression
Cell Death and Differentiation (2018) 25, 432–443 & 2018 ADMC Associazione Differenziamento e Morte Cellulare All rights reserved 1350-9047/18 www.nature.com/cdd Lin28B and miR-142-3p regulate neuronal differentiation by modulating Staufen1 expression Younseo Oh1,5, Jungyun Park1,5, Jin-Il Kim1, Mi-Yoon Chang2, Sang-Hun Lee3, Youl-Hee Cho*,4 and Jungwook Hwang*,1,4 Staufen1 (STAU1) and Lin28B are RNA-binding proteins that are involved in neuronal differentiation as a function of post- transcriptional regulation. STAU1 triggers post-transcriptional regulation, including mRNA export, mRNA relocation, translation and mRNA decay. Lin28B also has multiple functions in miRNA biogenesis and the regulation of translation. Here, we examined the connection between STAU1 and Lin28B and found that Lin28B regulates the abundance of STAU1 mRNA via miRNA maturation. Decreases in the expression of both STAU1 and Lin28B were observed during neuronal differentiation. Depletion of STAU1 or Lin28B inhibited neuronal differentiation, and overexpression of STAU1 or Lin28B enhanced neuronal differentiation. Interestingly, the stability of STAU1 mRNA was modulated by miR-142-3p, whose maturation was regulated by Lin28B. Thus, miR-142-3p expression increased as Lin28B expression decreased during differentiation, leading to the reduction of STAU1 expression. The transcriptome from Staufen-mediated mRNA decay (SMD) targets during differentiation was analyzed, confirming that STAU1 was a key factor in neuronal differentiation. In support of this finding, regulation of STAU1 expression in mouse neural precursor cells had the same effects on neuronal differentiation as it did in human neuroblastoma cells. These results revealed the collaboration of two RNA-binding proteins, STAU1 and Lin28B, as a regulatory mechanism in neuronal differentiation. -

Ribosomes Slide on Lysine-Encoding Homopolymeric a Stretches
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Crossref RESEARCH ARTICLE elifesciences.org Ribosomes slide on lysine-encoding homopolymeric A stretches Kristin S Koutmou1, Anthony P Schuller1, Julie L Brunelle1,2, Aditya Radhakrishnan1, Sergej Djuranovic3, Rachel Green1,2* 1Department of Molecular Biology and Genetics, Johns Hopkins School of Medicine, Baltimore, United States; 2Howard Hughes Medical Institute, Johns Hopkins School of Medicine, Baltimore, United States; 3Department of Cell Biology and Physiology, Washington University School of Medicine, St. Louis, United States Abstract Protein output from synonymous codons is thought to be equivalent if appropriate tRNAs are sufficiently abundant. Here we show that mRNAs encoding iterated lysine codons, AAA or AAG, differentially impact protein synthesis: insertion of iterated AAA codons into an ORF diminishes protein expression more than insertion of synonymous AAG codons. Kinetic studies in E. coli reveal that differential protein production results from pausing on consecutive AAA-lysines followed by ribosome sliding on homopolymeric A sequence. Translation in a cell-free expression system demonstrates that diminished output from AAA-codon-containing reporters results from premature translation termination on out of frame stop codons following ribosome sliding. In eukaryotes, these premature termination events target the mRNAs for Nonsense-Mediated-Decay (NMD). The finding that ribosomes slide on homopolymeric A sequences explains bioinformatic analyses indicating that consecutive AAA codons are under-represented in gene-coding sequences. Ribosome ‘sliding’ represents an unexpected type of ribosome movement possible during translation. DOI: 10.7554/eLife.05534.001 *For correspondence: ragreen@ Introduction jhmi.edu Messenger RNA (mRNA) transcripts can contain errors that result in the production of incorrect protein products. -
Primepcr™Assay Validation Report
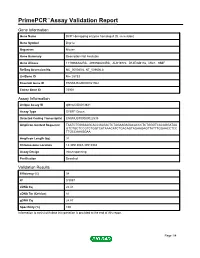
PrimePCR™Assay Validation Report Gene Information Gene Name DCP1 decapping enzyme homolog A (S. cerevisiae) Gene Symbol Dcp1a Organism Mouse Gene Summary Description Not Available Gene Aliases 1110066A22Rik, 4930568L04Rik, AU019772, D14Ertd817e, Mitc1, SMIF RefSeq Accession No. NC_000080.6, NT_039606.8 UniGene ID Mm.28733 Ensembl Gene ID ENSMUSG00000021962 Entrez Gene ID 75901 Assay Information Unique Assay ID qMmuCID0013841 Assay Type SYBR® Green Detected Coding Transcript(s) ENSMUST00000022535 Amplicon Context Sequence TAATCTGGGAAGCACCGAGACTCTAGAAGAGACACCCTCTGGGTCACAGGATAA GTCTGCTCCGTCTGGTCATAAACATCTGACAGTAGAAGAGTTATTTGGAACCTCC TTGCCAAAGGAA Amplicon Length (bp) 91 Chromosome Location 14:30513043-30518984 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 98 R2 0.9997 cDNA Cq 22.41 cDNA Tm (Celsius) 81 gDNA Cq 24.87 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Dcp1a, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Exon Amplification: a Strategy to Isolate Mammalian Genes Based on RNA Splicing (Gene Cloning/Polymerase Chain Reaction) ALAN J
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 4005-4009, May 1991 Genetics Exon amplification: A strategy to isolate mammalian genes based on RNA splicing (gene cloning/polymerase chain reaction) ALAN J. BUCKLER*, DAVID D. CHANG, SHARON L. GRAw, J. DAVID BROOK, DANIEL A. HABER, PHILLIP A. SHARP, AND DAVID E. HOUSMAN Center for Cancer Research, Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139 Contributed by Phillip A. Sharp, January 25, 1991 ABSTRACT We have developed a method, exon amplifi- (7, 8). Thus, this method may be generally applicable for the cation, for fast and efficient isolation of coding sequences from selection of exon sequences from any gene. The method is complex mammalian genomic DNA. This method is based on also both rapid and easily adapted to large scale experiments. the selection of RNA sequences, exons, which are flanked by A series of cloned genomic DNA fragments can be screened functional 5' and 3' splice sites. Fragments of cloned genomic within 1-2 weeks. The sensitivity of this method is high. DNA are inserted into an intron, which is flanked by 5' and 3' Genomic DNA segments of 20 kilobases (kb) or more can be splice sites of the human immunodeficiency virus 1 tat gene successfully screened in a single transfection by using a set contained within the plasmid pSPL1. COS-7 cells are trans- of pooled subclones. This method thus allows the rapid fected with these constructs, and the resulting RNA transcripts identification of exons in mammalian genomic DNA and are processed in vivo. Splice sites of exons contained within the should facilitate the isolation of a wide spectrum of genes of inserted genomic fragment are paired with splice sites of the significance in physiology and development. -

RNA Components of the Spliceosome Regulate Tissue- and Cancer-Specific Alternative Splicing
Downloaded from genome.cshlp.org on September 29, 2021 - Published by Cold Spring Harbor Laboratory Press Research RNA components of the spliceosome regulate tissue- and cancer-specific alternative splicing Heidi Dvinge,1,2,4 Jamie Guenthoer,3 Peggy L. Porter,3 and Robert K. Bradley1,2 1Computational Biology Program, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 2Basic Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA; 3Human Biology Division, Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, USA Alternative splicing of pre-mRNAs plays a pivotal role during the establishment and maintenance of human cell types. Characterizing the trans-acting regulatory proteins that control alternative splicing has therefore been the focus of much research. Recent work has established that even core protein components of the spliceosome, which are required for splicing to proceed, can nonetheless contribute to splicing regulation by modulating splice site choice. We here show that the RNA components of the spliceosome likewise influence alternative splicing decisions. Although these small nuclear RNAs (snRNAs), termed U1, U2, U4, U5, and U6 snRNA, are present in equal stoichiometry within the spliceosome, we found that their relative levels vary by an order of magnitude during development, across tissues, and across cancer samples. Physiologically relevant perturbation of individual snRNAs drove widespread gene-specific differences in alternative splic- ing but not transcriptome-wide splicing failure. Genes that were particularly sensitive to variations in snRNA abundance in a breast cancer cell line model were likewise preferentially misspliced within a clinically diverse cohort of invasive breast ductal carcinomas. -

Nonsense Mediated Mrna Decay As a Tool for Gene Therapy and the Role of Human DIS3L2 in Transcript Degradation
UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS DEPARTAMENTO DE BIOLOGIA ANIMAL mRNA Metabolism: Nonsense Mediated mRNA Decay as a Tool for Gene Therapy and the Role of Human DIS3L2 in Transcript Degradation Mestrado em Biologia Humana e Ambiente Gerson Leonel Asper Amaral Dissertação orientada por: Doutora Luísa Romão Professora Doutora Deodália Dias 2016 II UNIVERSIDADE DE LISBOA FACULDADE DE CIÊNCIAS DEPARTAMENTO DE BIOLOGIA ANIMAL mRNA Metabolism: Nonsense Mediated mRNA Decay as a Tool for Gene Therapy and the Role of Human DIS3L2 in Transcript Degradation Mestrado em Biologia Humana e Ambiente Gerson Leonel Asper Amaral Dissertação orientada por: Doutora Luísa Romão (Instituto Nacional de Saúde Dr. Ricardo Jorge) Professora Doutora Deodália Dias (Departamento de Biologia Animal, Faculdade de Ciências da Universidade de Lisboa) 2016 III IV “It is finished.” – Jesus Christ (The Bible, John 19:30) V VI ACKNOWLEDGEMENTS _______________________________________________________________________ This dissertation is the result of the very hard work, patience and resources from a lot of people. They were instrumental in the accomplishment of this project, be it through their knowledge, plain lab work, their friendship, guidance, support or sheer trust. I am sincerely thankful that all of you made part of my life at least for this year, because without you this would never see the light of day and would remain in the darkness of night. Clichéd poetry aside, honestly, thank you all. I want to start by thanking my main advisor, Dr. Luísa Romão, for accepting me into her brilliant lab and trusting me and my work. Thank you for sharing your vast knowledge with me, helping me, guiding me, being patient and calling my attention to my mistakes, all this without ever stopping from being pleasant! I feel so honoured and thankful. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Mrna Surveillance in Eukaryotes: Kinetic Proofreading of Proper Translation Termination As Assessed by Mrnp Domain Organization?
Downloaded from rnajournal.cshlp.org on October 5, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1999), 5:711–719+ Cambridge University Press+ Printed in the USA+ Copyright © 1999 RNA Society+ REVIEW mRNA surveillance in eukaryotes: Kinetic proofreading of proper translation termination as assessed by mRNP domain organization? PATRICIA HILLEREN and ROY PARKER Department of Molecular and Cellular Biology, Howard Hughes Medical Institute, University of Arizona, Tucson, Arizona 85721, USA ABSTRACT In the last few years it has become clear that a conserved mRNA degradation system, referred to as mRNA surveil- lance, exists in eukaryotic cells to degrade aberrant mRNAs. This process plays an important role in checking that mRNAs have been properly synthesized and functions, at least in part, to increase the fidelity of gene expression by degrading aberrant mRNAs that, if translated, would produce truncated proteins. A critical issue is how normal and aberrant mRNAs are distinguished and how that distinction leads to differences in mRNA stability. Recent results suggest a model with three main points. First, mRNPs have a domain organization that is, in part, a reflection of the completion of nuclear pre-mRNA processing events. Second, the critical aspect of distinguishing a normal from an aberrant mRNA is the environment of the translation termination codon as determined by the organization of the mRNP domains. Third, the cell distinguishes proper from improper termination through an internal clock that is the rate of ATP hydrolysis by Upf1p. If termination is completed before ATP hydrolysis, the mRNA is protected from mRNA degradation. Conversely, if termination is slow, then ATP hydrolysis and a structural rearrangement occurs before termination is completed, which affects the fate of the terminating ribosome in a manner that fails to stabilize the mRNA. -

Sequences at the Exon-Intron Boundaries* (Split Gene/Mrna Splicing/Eukaryotic Gene Structure) R
Proc. Nati. Acad. Sci. USA Vol. 75, No. 10, pp. 4853-4857, October 1978 Biochemistry Ovalbumin gene: Evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries* (split gene/mRNA splicing/eukaryotic gene structure) R. BREATHNACH, C. BENOIST, K. O'HARE, F. GANNON, AND P. CHAMBON Laboratoire de Genetique Mol6culaire des Eucaryotes du Centre National de la Recherche Scientifique, Unite 44 de l'Institut National de la Sant6 et de la Recherche MWdicale, Institut de Chimie Biologique, Facult6 de Melecine, Strasbourg 67085, France Communicated by A. Frey-Wyssling, July 31, 1978 ABSTRACT Selected regions of cloned EcoRI fragments the 5' end of ov-mRNA and have revealed some interesting of the chicken ovalbumin gene have been sequenced. The po- features in the DNA sequences at exon-intron boundaries. sitions where the sequences coding for ovalbumin mRNA (ov- mRNA) are interrupted in the genome have been determined, and a previously unreported interruption in the DNA sequences MATERIALS AND METHODS coding for the 5' nontranslated region of the messenger has been discovered. Because directly repeated sequences are found at Plasmid pCR1 ov 2.1 containing the ov-ds-cDNA insert was exon-intron boundaries, the nucleotide sequence alone cannot prepared as described (9). EcoRI fragments "b," "c," and "d" define unique excision-ligation points for the processing of a previously cloned in X vectors (3) were transferred to the plas- possible ov-mRNA precursor. However, the sequences in these mid pBR 322. An EcoRI/HindIII of the EcoRI boundary regions share common features; this leads to the fragment proposal that there are, in fact, unique excision-ligation points fragment "a" containing the entirety of exon 7 (Fig. -

Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins
Biomolecules 2015, 5, 1441-1466; doi:10.3390/biom5031441 OPEN ACCESS biomolecules ISSN 2218-273X www.mdpi.com/journal/biomolecules/ Article Comprehensive Protein Interactome Analysis of a Key RNA Helicase: Detection of Novel Stress Granule Proteins Rebecca Bish 1,†, Nerea Cuevas-Polo 1,†, Zhe Cheng 1, Dolores Hambardzumyan 2, Mathias Munschauer 3, Markus Landthaler 3 and Christine Vogel 1,* 1 Center for Genomics and Systems Biology, Department of Biology, New York University, 12 Waverly Place, New York, NY 10003, USA; E-Mails: [email protected] (R.B.); [email protected] (N.C.-P.); [email protected] (Z.C.) 2 The Cleveland Clinic, Department of Neurosciences, Lerner Research Institute, 9500 Euclid Avenue, Cleveland, OH 44195, USA; E-Mail: [email protected] 3 RNA Biology and Post-Transcriptional Regulation, Max-Delbrück-Center for Molecular Medicine, Berlin-Buch, Robert-Rössle-Str. 10, Berlin 13092, Germany; E-Mails: [email protected] (M.M.); [email protected] (M.L.) † These authors contributed equally to this work. * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-212-998-3976; Fax: +1-212-995-4015. Academic Editor: André P. Gerber Received: 10 May 2015 / Accepted: 15 June 2015 / Published: 15 July 2015 Abstract: DDX6 (p54/RCK) is a human RNA helicase with central roles in mRNA decay and translation repression. To help our understanding of how DDX6 performs these multiple functions, we conducted the first unbiased, large-scale study to map the DDX6-centric protein-protein interactome using immunoprecipitation and mass spectrometry. Using DDX6 as bait, we identify a high-confidence and high-quality set of protein interaction partners which are enriched for functions in RNA metabolism and ribosomal proteins.