1 Whole Genome Sequencing and Rare Variant Analysis in Essential
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

PRODUCT SPECIFICATION Product Datasheet
Product Datasheet QPrEST PRODUCT SPECIFICATION Product Name QPrEST SLIT3 Mass Spectrometry Protein Standard Product Number QPrEST21571 Protein Name Slit homolog 3 protein Uniprot ID O75094 Gene SLIT3 Product Description Stable isotope-labeled standard for absolute protein quantification of Slit homolog 3 protein. Lys (13C and 15N) and Arg (13C and 15N) metabolically labeled recombinant human protein fragment. Application Absolute protein quantification using mass spectrometry Sequence (excluding CDNKNDSANACSAFKCHHGQCHISDQGEPYCLCQPGFSGEHCQQENPCLG fusion tag) QVVREVIRRQKGYASCATASKVPIMECRG Theoretical MW 26448 Da including N-terminal His6ABP fusion tag Fusion Tag A purification and quantification tag (QTag) consisting of a hexahistidine sequence followed by an Albumin Binding Protein (ABP) domain derived from Streptococcal Protein G. Expression Host Escherichia coli LysA ArgA BL21(DE3) Purification IMAC purification Purity >90% as determined by Bioanalyzer Protein 230 Purity Assay Isotopic Incorporation >99% Concentration >5 μM after reconstitution in 100 μl H20 Concentration Concentration determined by LC-MS/MS using a highly pure amino acid analyzed internal Determination reference (QTag), CV ≤10%. Amount >0.5 nmol per vial, two vials supplied. Formulation Lyophilized in 100 mM Tris-HCl 5% Trehalose, pH 8.0 Instructions for Spin vial before opening. Add 100 μL ultrapure H2O to the vial. Vortex thoroughly and spin Reconstitution down. For further dilution, see Application Protocol. Shipping Shipped at ambient temperature Storage Lyophilized product shall be stored at -20°C. See COA for expiry date. Reconstituted product can be stored at -20°C for up to 4 weeks. Avoid repeated freeze-thaw cycles. Notes For research use only Product of Sweden. For research use only. Not intended for pharmaceutical development, diagnostic, therapeutic or any in vivo use. -

Versican V2 Assembles the Extracellular Matrix Surrounding the Nodes of Ranvier in the CNS
The Journal of Neuroscience, June 17, 2009 • 29(24):7731–7742 • 7731 Cellular/Molecular Versican V2 Assembles the Extracellular Matrix Surrounding the Nodes of Ranvier in the CNS María T. Dours-Zimmermann,1 Konrad Maurer,2 Uwe Rauch,3 Wilhelm Stoffel,4 Reinhard Fa¨ssler,5 and Dieter R. Zimmermann1 Institutes of 1Surgical Pathology and 2Anesthesiology, University Hospital Zurich, CH-8091 Zurich, Switzerland, 3Vascular Wall Biology, Department of Experimental Medical Science, University of Lund, S-221 00 Lund, Sweden, 4Center for Biochemistry, Medical Faculty, University of Cologne, D-50931 Cologne, Germany, and 5Department of Molecular Medicine, Max Planck Institute of Biochemistry, D-82152 Martinsried, Germany The CNS-restricted versican splice-variant V2 is a large chondroitin sulfate proteoglycan incorporated in the extracellular matrix sur- rounding myelinated fibers and particularly accumulating at nodes of Ranvier. In vitro, it is a potent inhibitor of axonal growth and therefore considered to participate in the reduction of structural plasticity connected to myelination. To study the role of versican V2 during postnatal development, we designed a novel isoform-specific gene inactivation approach circumventing early embryonic lethality of the complete knock-out and preventing compensation by the remaining versican splice variants. These mice are viable and fertile; however, they display major molecular alterations at the nodes of Ranvier. While the clustering of nodal sodium channels and paranodal structures appear in versican V2-deficient mice unaffected, the formation of the extracellular matrix surrounding the nodes is largely impaired. The conjoint loss of tenascin-R and phosphacan from the perinodal matrix provide strong evidence that versican V2, possibly controlled by a nodal receptor, organizes the extracellular matrix assembly in vivo. -

Supplementary Materials: Evaluation of Cytotoxicity and Α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids
Supplementary Materials: Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids Oxana B. Kazakova1*, Gul'nara V. Giniyatullina1, Akhat G. Mustafin1, Denis A. Babkov2, Elena V. Sokolova2, Alexander A. Spasov2* 1Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences, 71, pr. Oktyabrya, 450054 Ufa, Russian Federation 2Scientific Center for Innovative Drugs, Volgograd State Medical University, Novorossiyskaya st. 39, Volgograd 400087, Russian Federation Correspondence Prof. Dr. Oxana B. Kazakova Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences 71 Prospeсt Oktyabrya Ufa, 450054 Russian Federation E-mail: [email protected] Prof. Dr. Alexander A. Spasov Scientific Center for Innovative Drugs of the Volgograd State Medical University 39 Novorossiyskaya st. Volgograd, 400087 Russian Federation E-mail: [email protected] Figure S1. 1H and 13C of compound 2. H NH N H O H O H 2 2 Figure S2. 1H and 13C of compound 4. NH2 O H O H CH3 O O H H3C O H 4 3 Figure S3. Anticancer screening data of compound 2 at single dose assay 4 Figure S4. Anticancer screening data of compound 7 at single dose assay 5 Figure S5. Anticancer screening data of compound 8 at single dose assay 6 Figure S6. Anticancer screening data of compound 9 at single dose assay 7 Figure S7. Anticancer screening data of compound 12 at single dose assay 8 Figure S8. Anticancer screening data of compound 13 at single dose assay 9 Figure S9. Anticancer screening data of compound 14 at single dose assay 10 Figure S10. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Kelch Proteins: Emerging Roles in Skeletal Muscle Development and Diseases
Kelch proteins: emerging roles in skeletal muscle development and diseases The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Gupta, Vandana A., and Alan H Beggs. 2014. “Kelch proteins: emerging roles in skeletal muscle development and diseases.” Skeletal Muscle 4 (1): 11. doi:10.1186/2044-5040-4-11. http:// dx.doi.org/10.1186/2044-5040-4-11. Published Version doi:10.1186/2044-5040-4-11 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:12406733 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Gupta and Beggs Skeletal Muscle 2014, 4:11 http://www.skeletalmusclejournal.com/content/4/1/11 REVIEW Open Access Kelch proteins: emerging roles in skeletal muscle development and diseases Vandana A Gupta and Alan H Beggs* Abstract Our understanding of genes that cause skeletal muscle disease has increased tremendously over the past three decades. Advances in approaches to genetics and genomics have aided in the identification of new pathogenic mechanisms in rare genetic disorders and have opened up new avenues for therapeutic interventions by identification of new molecular pathways in muscle disease. Recent studies have identified mutations of several Kelch proteins in skeletal muscle disorders. The Kelch superfamily is one of the largest evolutionary conserved gene families. The 66 known family members all possess a Kelch-repeat containing domain and are implicated in diverse biological functions. -
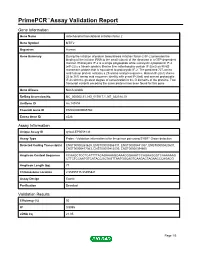
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mitochondrial translational initiation factor 2 Gene Symbol MTIF2 Organism Human Gene Summary During the initiation of protein biosynthesis initiation factor-2 (IF-2) promotes the binding of the initiator tRNA to the small subunit of the ribosome in a GTP-dependent manner. Prokaryotic IF-2 is a single polypeptide while eukaryotic cytoplasmic IF-2 (eIF-2) is a trimeric protein. Bovine liver mitochondria contain IF-2(mt) an 85-kD monomeric protein that is equivalent to prokaryotic IF-2. The predicted 727-amino acid human protein contains a 29-amino acid presequence. Human IF-2(mt) shares 32 to 38% amino acid sequence identity with yeast IF-2(mt) and several prokaryotic IF-2s with the greatest degree of conservation in the G domains of the proteins. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases Not Available RefSeq Accession No. NC_000002.11, NG_017017.1, NT_022184.15 UniGene ID Hs.149894 Ensembl Gene ID ENSG00000085760 Entrez Gene ID 4528 Assay Information Unique Assay ID qHsaCEP0058136 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000263629, ENST00000366137, ENST00000441307, ENST00000420637, ENST00000417363, ENST00000412530, ENST00000394600 Amplicon Context Sequence CCAAGCTCCTCATTTTACAGAAAAGGAAACGGAAATCCAGAAGGGTCAAAAAAG CTTCTCCAATGTCATACCGCTAGTTAATGGCAGTCAAGACTAGAACCCAGACG Amplicon Length (bp) 77 Chromosome Location 2:55495715-55495821 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9995 cDNA Cq 21.05 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.56 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Upregulation of Peroxisome Proliferator-Activated Receptor-Α And
Upregulation of peroxisome proliferator-activated receptor-α and the lipid metabolism pathway promotes carcinogenesis of ampullary cancer Chih-Yang Wang, Ying-Jui Chao, Yi-Ling Chen, Tzu-Wen Wang, Nam Nhut Phan, Hui-Ping Hsu, Yan-Shen Shan, Ming-Derg Lai 1 Supplementary Table 1. Demographics and clinical outcomes of five patients with ampullary cancer Time of Tumor Time to Age Differentia survival/ Sex Staging size Morphology Recurrence recurrence Condition (years) tion expired (cm) (months) (months) T2N0, 51 F 211 Polypoid Unknown No -- Survived 193 stage Ib T2N0, 2.41.5 58 F Mixed Good Yes 14 Expired 17 stage Ib 0.6 T3N0, 4.53.5 68 M Polypoid Good No -- Survived 162 stage IIA 1.2 T3N0, 66 M 110.8 Ulcerative Good Yes 64 Expired 227 stage IIA T3N0, 60 M 21.81 Mixed Moderate Yes 5.6 Expired 16.7 stage IIA 2 Supplementary Table 2. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of an ampullary cancer microarray using the Database for Annotation, Visualization and Integrated Discovery (DAVID). This table contains only pathways with p values that ranged 0.0001~0.05. KEGG Pathway p value Genes Pentose and 1.50E-04 UGT1A6, CRYL1, UGT1A8, AKR1B1, UGT2B11, UGT2A3, glucuronate UGT2B10, UGT2B7, XYLB interconversions Drug metabolism 1.63E-04 CYP3A4, XDH, UGT1A6, CYP3A5, CES2, CYP3A7, UGT1A8, NAT2, UGT2B11, DPYD, UGT2A3, UGT2B10, UGT2B7 Maturity-onset 2.43E-04 HNF1A, HNF4A, SLC2A2, PKLR, NEUROD1, HNF4G, diabetes of the PDX1, NR5A2, NKX2-2 young Starch and sucrose 6.03E-04 GBA3, UGT1A6, G6PC, UGT1A8, ENPP3, MGAM, SI, metabolism -

The Expression of the Human Apolipoprotein Genes and Their Regulation by Ppars
CORE Metadata, citation and similar papers at core.ac.uk Provided by UEF Electronic Publications The expression of the human apolipoprotein genes and their regulation by PPARs Juuso Uski M.Sc. Thesis Biochemistry Department of Biosciences University of Kuopio June 2008 Abstract The expression of the human apolipoprotein genes and their regulation by PPARs. UNIVERSITY OF KUOPIO, the Faculty of Natural and Environmental Sciences, Curriculum of Biochemistry USKI Juuso Oskari Thesis for Master of Science degree Supervisors Prof. Carsten Carlberg, Ph.D. Merja Heinäniemi, Ph.D. June 2008 Keywords: nuclear receptors; peroxisome proliferator-activated receptor; PPAR response element; apolipoprotein; lipid metabolism; high density lipoprotein; low density lipoprotein. Lipids are any fat-soluble, naturally-occurring molecules and one of their main biological functions is energy storage. Lipoproteins carry hydrophobic lipids in the water and salt-based blood environment for processing and energy supply in liver and other organs. In this study, the genomic area around the apolipoprotein genes was scanned in silico for PPAR response elements (PPREs) using the in vitro data-based computer program. Several new putative REs were found in surroundings of multiple lipoprotein genes. The responsiveness of those apolipoprotein genes to the PPAR ligands GW501516, rosiglitazone and GW7647 in the HepG2, HEK293 and THP-1 cell lines were tested with real-time PCR. The APOA1, APOA2, APOB, APOD, APOE, APOF, APOL1, APOL3, APOL5 and APOL6 genes were found to be regulated by PPARs in direct or secondary manners. Those results provide new insights in the understanding of lipid metabolism and so many lifestyle diseases like atherosclerosis, type 2 diabetes, heart disease and stroke. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Origin and Evolution of Human Ampliconic Gene Families and Ampliconic Structure
Downloaded from genome.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press Letter The origin and evolution of human ampliconic gene families and ampliconic structure Bejon Kumar Bhowmick, Yoko Satta, and Naoyuki Takahata1 Department of Biosystems Science, The Graduate University for Advance Studies (Sokendai), Kanagawa 240-0193, Japan Out of the nine male-specific gene families in the human Y chromosome amplicons, we investigate the origin and evolution of seven families for which gametologous and orthologous sequences are available. Proto-X/Y gene pairs in the original mammalian sex chromosomes played major roles in origins and gave rise to five gene families: XKRY, VCY, HSFY, RBMY, and TSPY. The divergence times between gametologous X- and Y-linked copies in these families are well correlated with the former X-chromosomal locations. The CDY and DAZ families originated exceptionally by retroposition and transposition of autosomal copies, respectively, but CDY possesses an X-linked copy of enigmatic origin. We also investigate the evolutionary relatedness among Y-linked copies of a gene family in light of their ampliconic locations (palindromes, inverted repeats, and the TSPY array). Although any pair of copies located at the same arm positions within a palindrome is identical or nearly so by frequent gene conversion, copies located at different arm positions are distinctively different. Since these and other distinct copies in various gene families were amplified almost simultaneously in the stem lineage of Catarrhini, we take these simultaneous amplifications as evidence for the elaborate formation of Y ampliconic structure. Curiously, some copies in a gene family located at different palindromes exhibit high sequence similarity, and in most cases, such similarity greatly extends to repeat units that harbor these copies. -

1 CETP Inhibition Improves HDL Function but Leads to Fatty Liver and Insulin Resistance in CETP-Expressing Transgenic Mice on A
Page 1 of 55 Diabetes CETP inhibition improves HDL function but leads to fatty liver and insulin resistance in CETP-expressing transgenic mice on a high-fat diet Lin Zhu1,2, Thao Luu2, Christopher H. Emfinger1,2, Bryan A Parks5, Jeanne Shi2,7, Elijah Trefts3, Fenghua Zeng4, Zsuzsanna Kuklenyik5, Raymond C. Harris4, David H. Wasserman3, Sergio Fazio6 and John M. Stafford1,2,3,* 1VA Tennessee Valley Healthcare System, 2Division of Diabetes, Endocrinology, & Metabolism, 3Department of Molecular Physiology and Biophysics, 4Devision of Nephrology and Hypertension, Vanderbilt University School of Medicine. 5Division of Laboratory Sciences, Centers for Disease Control and Prevention. 6The Center for Preventive Cardiology at the Knight Cardiovascular Institute, Oregon Health & Science University. 7Trinity College of Art and Science, Duke University. * Address correspondence and request for reprints to: John. M. Stafford, 7445D Medical Research Building IV, Nashville, TN 37232-0475, phone (615) 936-6113, fax (615) 936- 1667 Email: [email protected] Running Title: CETP inhibition and insulin resistance Word Count: 5439 Figures: 7 Tables: 1 1 Diabetes Publish Ahead of Print, published online September 13, 2018 Diabetes Page 2 of 55 Abstract In clinical trials inhibition of cholesteryl ester transfer protein (CETP) raises HDL cholesterol levels but doesn’t robustly improve cardiovascular outcomes. About 2/3 of trial participants were obese. Lower plasma CETP activity is associated with increased cardiovascular risk in human studies, and protective aspects of CETP have been observed in mice fed a high-fat diet (HFD) with regard to metabolic outcomes. To define if CETP inhibition has different effects depending on the presence of obesity, we performed short- term anacetrapib treatment in chow- and HFD-fed CETP-transgenic mice.