NIH Public Access Author Manuscript Biochim Biophys Acta
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
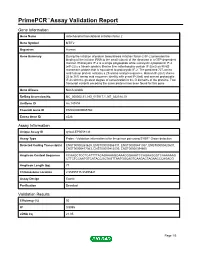
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name mitochondrial translational initiation factor 2 Gene Symbol MTIF2 Organism Human Gene Summary During the initiation of protein biosynthesis initiation factor-2 (IF-2) promotes the binding of the initiator tRNA to the small subunit of the ribosome in a GTP-dependent manner. Prokaryotic IF-2 is a single polypeptide while eukaryotic cytoplasmic IF-2 (eIF-2) is a trimeric protein. Bovine liver mitochondria contain IF-2(mt) an 85-kD monomeric protein that is equivalent to prokaryotic IF-2. The predicted 727-amino acid human protein contains a 29-amino acid presequence. Human IF-2(mt) shares 32 to 38% amino acid sequence identity with yeast IF-2(mt) and several prokaryotic IF-2s with the greatest degree of conservation in the G domains of the proteins. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases Not Available RefSeq Accession No. NC_000002.11, NG_017017.1, NT_022184.15 UniGene ID Hs.149894 Ensembl Gene ID ENSG00000085760 Entrez Gene ID 4528 Assay Information Unique Assay ID qHsaCEP0058136 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000263629, ENST00000366137, ENST00000441307, ENST00000420637, ENST00000417363, ENST00000412530, ENST00000394600 Amplicon Context Sequence CCAAGCTCCTCATTTTACAGAAAAGGAAACGGAAATCCAGAAGGGTCAAAAAAG CTTCTCCAATGTCATACCGCTAGTTAATGGCAGTCAAGACTAGAACCCAGACG Amplicon Length (bp) 77 Chromosome Location 2:55495715-55495821 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9995 cDNA Cq 21.05 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.56 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Role of Phytochemicals in Colon Cancer Prevention: a Nutrigenomics Approach
Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan J van Erk Promotor: Prof. Dr. P.J. van Bladeren Hoogleraar in de Toxicokinetiek en Biotransformatie Wageningen Universiteit Co-promotoren: Dr. Ir. J.M.M.J.G. Aarts Universitair Docent, Sectie Toxicologie Wageningen Universiteit Dr. Ir. B. van Ommen Senior Research Fellow Nutritional Systems Biology TNO Voeding, Zeist Promotiecommissie: Prof. Dr. P. Dolara University of Florence, Italy Prof. Dr. J.A.M. Leunissen Wageningen Universiteit Prof. Dr. J.C. Mathers University of Newcastle, United Kingdom Prof. Dr. M. Müller Wageningen Universiteit Dit onderzoek is uitgevoerd binnen de onderzoekschool VLAG Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan Jolanda van Erk Proefschrift ter verkrijging van graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof.Dr.Ir. L. Speelman, in het openbaar te verdedigen op vrijdag 1 oktober 2004 des namiddags te vier uur in de Aula Title Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Author Marjan Jolanda van Erk Thesis Wageningen University, Wageningen, the Netherlands (2004) with abstract, with references, with summary in Dutch ISBN 90-8504-085-X ABSTRACT Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Specific food compounds, especially from fruits and vegetables, may protect against development of colon cancer. In this thesis effects and mechanisms of various phytochemicals in relation to colon cancer prevention were studied through application of large-scale gene expression profiling. Expression measurement of thousands of genes can yield a more complete and in-depth insight into the mode of action of the compounds. -

By Submitted in Partial Satisfaction of the Requirements for Degree of in In
BCL6 maintains thermogenic capacity of brown adipose tissue during dormancy by Vassily Kutyavin DISSERTATION Submitted in partial satisfaction of the requirements for degree of DOCTOR OF PHILOSOPHY in Biomedical Sciences in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________Eric Verdin Chair ______________________________________________________________________________Ajay Chawla ______________________________________________________________________________Ethan Weiss ______________________________________________________________________________ ______________________________________________________________________________ Committee Members Copyright 2019 by Vassily Kutyavin ii Dedicated to everyone who has supported me during my scientific education iii Acknowledgements I'm very grateful to my thesis adviser, Ajay Chawla, for his mentorship and support during my dissertation work over the past five years. Throughout my time in his lab, I was always able to rely on his guidance, and his enthusiasm for science was a great source of motivation. Even when he was traveling, he could easily be reached for advice by phone or e- mail. I am particularly grateful for his help with writing the manuscript, which was probably the most challenging aspect of graduate school for me. I am also very grateful to him for helping me find a postdoctoral fellowship position. Ajay's inquisitive and fearless approach to science have been a great inspiration to me. In contrast to the majority of scientists who focus narrowly on a specific topic, Ajay pursued fundamental questions across a broad range of topics and was able to make tremendous contributions. My experience in his lab instilled in me a deep appreciation for thinking about the entire organism from an evolutionary perspective and focusing on the key questions that escape the attention of the larger scientific community. As I move forward in my scientific career, there is no doubt that I will rely on him as a role model. -

Stroke Genetics and Genomics
Faculdade de Medicina da Universidade de Lisboa Unidade Neurológica de Investigação Clínica PhD Thesis Stroke Genetics and Genomics Tiago Krug Coelho Host Institution: Instituto Gulbenkian de Ciência Supervisor at Instituto Gulbenkian de Ciência: Doctor Sofia Oliveira Supervisor at Faculdade de Medicina da Universidade de Lisboa: Professor José Ferro PhD in Biomedical Sciences Specialization in Neurosciences 2010 Stroke Genetics and Genomics A ciência tem, de facto, um único objectivo: a verdade. Não esgota perfeitamente a sua tarefa se não descobre a causa do todo. Chiara Lubich i Stroke Genetics and Genomics ii Stroke Genetics and Genomics A impressão desta dissertação foi aprovada pela Comissão Coordenadora do Conselho Científico da Faculdade de Medicina de Lisboa em reunião de 28 de Setembro de 2010. iii Stroke Genetics and Genomics iv Stroke Genetics and Genomics As opiniões expressas são da exclusiva responsabilidade do seu autor. v Stroke Genetics and Genomics vi Stroke Genetics and Genomics Abstract ABSTRACT This project presents a comprehensive approach to the identification of new genes that influence the risk for developing stroke. Stroke is the leading cause of death in Portugal and the third leading cause of death in the developed world. It is even more disabling than lethal, and the persistent neurological impairment and physical disability caused by stroke have a very high socioeconomic cost. Moreover, the number of affected individuals is expected to increase with the current aging of the population. Stroke is a “brain attack” cutting off vital blood and oxygen to the brain cells and it is a complex disease resulting from environmental and genetic factors. -

Mitochondrial Genetics
Mitochondrial genetics Patrick Francis Chinnery and Gavin Hudson* Institute of Genetic Medicine, International Centre for Life, Newcastle University, Central Parkway, Newcastle upon Tyne NE1 3BZ, UK Introduction: In the last 10 years the field of mitochondrial genetics has widened, shifting the focus from rare sporadic, metabolic disease to the effects of mitochondrial DNA (mtDNA) variation in a growing spectrum of human disease. The aim of this review is to guide the reader through some key concepts regarding mitochondria before introducing both classic and emerging mitochondrial disorders. Sources of data: In this article, a review of the current mitochondrial genetics literature was conducted using PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). In addition, this review makes use of a growing number of publically available databases including MITOMAP, a human mitochondrial genome database (www.mitomap.org), the Human DNA polymerase Gamma Mutation Database (http://tools.niehs.nih.gov/polg/) and PhyloTree.org (www.phylotree.org), a repository of global mtDNA variation. Areas of agreement: The disruption in cellular energy, resulting from defects in mtDNA or defects in the nuclear-encoded genes responsible for mitochondrial maintenance, manifests in a growing number of human diseases. Areas of controversy: The exact mechanisms which govern the inheritance of mtDNA are hotly debated. Growing points: Although still in the early stages, the development of in vitro genetic manipulation could see an end to the inheritance of the most severe mtDNA disease. Keywords: mitochondria/genetics/mitochondrial DNA/mitochondrial disease/ mtDNA Accepted: April 16, 2013 Mitochondria *Correspondence address. The mitochondrion is a highly specialized organelle, present in almost all Institute of Genetic Medicine, International eukaryotic cells and principally charged with the production of cellular Centre for Life, Newcastle energy through oxidative phosphorylation (OXPHOS). -

Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves
G C A T T A C G G C A T genes Article Plateau Grass and Greenhouse Flower? Distinct Genetic Basis of Closely Related Toad Tadpoles Respectively Adapted to High Altitude and Karst Caves 1,2, 1, 1,2 1,2 1, Liming Chang y, Wei Zhu y, Shengchao Shi , Meihua Zhang , Jianping Jiang *, Cheng Li 1, Feng Xie 1 and Bin Wang 1,* 1 CAS Key Laboratory of Mountain Ecological Restoration and Bioresource Utilization & Ecological Restoration and Biodiversity Conservation Key Laboratory of Sichuan Province, Chengdu Institute of Biology, Chinese Academy of Sciences, Chengdu 610041, China; [email protected] (L.C.); [email protected] (W.Z.); [email protected] (S.S.); [email protected] (M.Z.); [email protected] (C.L.); [email protected] (F.X.) 2 University of Chinese Academy of Sciences, Beijing 100049, China * Correspondence: [email protected] (B.W.); [email protected] (J.J.) These authors have contributed equally to this work. y Received: 1 December 2019; Accepted: 19 January 2020; Published: 22 January 2020 Abstract: Genetic adaptation to extremes is a fascinating topic. Nevertheless, few studies have explored the genetic adaptation of closely related species respectively inhabiting distinct extremes. With deep transcriptome sequencing, we attempt to detect the genetic architectures of tadpoles of five closely related toad species adapted to the Tibetan Plateau, middle-altitude mountains and karst caves. Molecular evolution analyses indicated that not only the number of fast evolving genes (FEGs), but also the functioning coverage of FEGs, increased with elevation. Enrichment analyses correspondingly revealed that the highland species had most of the FEGs involved in high-elevation adaptation, for example, amino acid substitutions of XRCC6 in its binding domains might improve the capacity of DNA repair of the toad. -

Report a Recessive Contiguous Gene Deletion of Chromosome 2P16
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Am. J. Hum. Genet. 69:869–875, 2001 Report A Recessive Contiguous Gene Deletion of Chromosome 2p16 Associated with Cystinuria and a Mitochondrial Disease Ruti Parvari,1 Irena Brodyansky,2 Orly Elpeleg,3 Shimon Moses,2 Daniel Landau,2 and Eli Hershkovitz2 1Department of Developmental Molecular Genetics, Faculty of Health Sciences, and 2Department of Pediatrics, Soroka Medical Center and Faculty of Health Sciences, Ben Gurion University of the Negev, Beer Sheva, Israel; and 3Metabolic Disease Unit, Shaare Zedek Medical Center, Jerusalem Deletions ranging from 100 Kb to 1 Mb—too small to be detected under the microscope—may still involve dozens of genes, thus causing microdeletion syndromes. The vast majority of these syndromes are caused by haploinsuf- ficiency of one or several genes and are transmitted as dominant traits. We identified seven patients originating from an extended family and presenting with a unique syndrome, inherited in a recessive mode, consisting of cystinuria, neonatal seizures, hypotonia, severe somatic and developmental delay, facial dysmorphism, and lactic acidemia. Reduced activity of all the respiratory chain enzymatic complexes that are encoded in the mitochondria was found in muscle biopsy specimens of the patients examined. The molecular basis of this disorder is a homozygous deletion of 179,311 bp on chromosome 2p16, which includes the type I cystinuria gene (SLC3A1), the protein phosphatase 2Cb gene (PP2Cb), an unidentified gene (KIAA0436), and several expressed sequence tags. The extent of the deletion suggests that this unique syndrome is related to the complete absence of these genes’ products, one of which may be essential for the synthesis of mitochondrial encoded proteins. -

Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations Fall 2010 Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Renuka Nayak University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Computational Biology Commons, and the Genomics Commons Recommended Citation Nayak, Renuka, "Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress" (2010). Publicly Accessible Penn Dissertations. 1559. https://repository.upenn.edu/edissertations/1559 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/1559 For more information, please contact [email protected]. Coexpression Networks Based on Natural Variation in Human Gene Expression at Baseline and Under Stress Abstract Genes interact in networks to orchestrate cellular processes. Here, we used coexpression networks based on natural variation in gene expression to study the functions and interactions of human genes. We asked how these networks change in response to stress. First, we studied human coexpression networks at baseline. We constructed networks by identifying correlations in expression levels of 8.9 million gene pairs in immortalized B cells from 295 individuals comprising three independent samples. The resulting networks allowed us to infer interactions between biological processes. We used the network to predict the functions of poorly-characterized human genes, and provided some experimental support. Examining genes implicated in disease, we found that IFIH1, a diabetes susceptibility gene, interacts with YES1, which affects glucose transport. Genes predisposing to the same diseases are clustered non-randomly in the network, suggesting that the network may be used to identify candidate genes that influence disease susceptibility. -

Systematic Detection of Brain Protein-Coding Genes Under Positive Selection During Primate Evolution and Their Roles in Cognition
Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Title: Systematic detection of brain protein-coding genes under positive selection during primate evolution and their roles in cognition Short title: Evolution of brain protein-coding genes in humans Guillaume Dumasa,b, Simon Malesysa, and Thomas Bourgerona a Human Genetics and Cognitive Functions, Institut Pasteur, UMR3571 CNRS, Université de Paris, Paris, (75015) France b Department of Psychiatry, Université de Montreal, CHU Ste Justine Hospital, Montreal, QC, Canada. Corresponding author: Guillaume Dumas Human Genetics and Cognitive Functions Institut Pasteur 75015 Paris, France Phone: +33 6 28 25 56 65 [email protected] Dumas, Malesys, and Bourgeron 1 of 40 Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Abstract The human brain differs from that of other primates, but the genetic basis of these differences remains unclear. We investigated the evolutionary pressures acting on almost all human protein-coding genes (N=11,667; 1:1 orthologs in primates) based on their divergence from those of early hominins, such as Neanderthals, and non-human primates. We confirm that genes encoding brain-related proteins are among the most strongly conserved protein-coding genes in the human genome. Combining our evolutionary pressure metrics for the protein- coding genome with recent datasets, we found that this conservation applied to genes functionally associated with the synapse and expressed in brain structures such as the prefrontal cortex and the cerebellum. Conversely, several genes presenting signatures commonly associated with positive selection appear as causing brain diseases or conditions, such as micro/macrocephaly, Joubert syndrome, dyslexia, and autism. -

Genome-Wide Association Study of Alcohol Dependence: Significant findings in African- and European-Americans Including Novel Risk Loci
Molecular Psychiatry (2014) 19, 41–49 & 2014 Macmillan Publishers Limited All rights reserved 1359-4184/14 www.nature.com/mp IMMEDIATE COMMUNICATION Genome-wide association study of alcohol dependence: significant findings in African- and European-Americans including novel risk loci J Gelernter1,2, HR Kranzler3, R Sherva4, L Almasy5, R Koesterer4, AH Smith1, R Anton6, UW Preuss7, M Ridinger8, D Rujescu7, N Wodarz8, P Zill9, H Zhao10,11 and LA Farrer4,12 We report a GWAS of alcohol dependence (AD) in European-American (EA) and African-American (AA) populations, with replication in independent samples of EAs, AAs and Germans. Our sample for discovery and replication was 16 087 subjects, the largest sample for AD GWAS to date. Numerous genome-wide significant (GWS) associations were identified, many novel. Most associations were population specific, but in several cases were GWS in EAs and AAs for different SNPs at the same locus,showing biological convergence across populations. We confirmed well-known risk loci mapped to alcohol-metabolizing enzyme genes, notably ADH1B (EAs: Arg48His, P ¼ 1.17 Â 10 À 31; AAs: Arg369Cys, P ¼ 6.33 Â 10 À 17) and ADH1C in AAs (Thr151Thr, P ¼ 4.94 Â 10 À 10), and identified novel risk loci mapping to the ADH gene cluster on chromosome 4 and extending centromerically beyond it to include GWS associations at LOC100507053 in AAs (P ¼ 2.63 Â 10 À 11), PDLIM5 in EAs (P ¼ 2.01 Â 10 À 8), and METAP in AAs (P ¼ 3.35 Â 10 À 8). We also identified a novel GWS association (1.17 Â 10 À 10) mapped to chromosome 2 at rs1437396, between MTIF2 and CCDC88A, across all of the EA and AA cohorts, with supportive gene expression evidence, and population-specific GWS for markers on chromosomes 5, 9 and 19. -

1 Whole Genome Sequencing and Rare Variant Analysis in Essential
bioRxiv preprint doi: https://doi.org/10.1101/248443; this version posted January 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 2 Whole Genome Sequencing and Rare Variant Analysis in Essential Tremor 3 Families 4 5 Zagaa Odgerel1, Nora Hernandez2, Jemin Park2, Ruth Ottman3,4,5,6, Elan D. Louis2 and 6 Lorraine N. Clark1,7 7 8 9 1Department of Pathology and Cell Biology, College of Physicians and Surgeons, 10 Columbia University New York NY 10032 USA. 11 2Department of Neurology, Yale School of Medicine, Yale University, New Haven, CT 12 06510, USA; Department of Chronic Disease Epidemiology, Yale School of Public 13 Health, New Haven, CT 06510, USA. 14 3G.H Sergievsky Center, Columbia University, New York, NY 10032 USA. 15 4Department of Neurology, College of Physicians and Surgeons, Columbia University 16 New York, NY 10032 USA. 17 5Department of Epidemiology, Mailman School of Public Health, Columbia University, 18 NY 10032 USA. 19 6Division of Epidemiology, New York State Psychiatric Institute, New York NY 10032 20 USA. 21 7Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, College of 22 Physicians and Surgeons, Columbia University, New York, NY 10032 USA. 23 24 25 26 1 bioRxiv preprint doi: https://doi.org/10.1101/248443; this version posted January 16, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. -

Transcriptomic and Proteomic Analysis of Human Hepatic Stellate Cells Treated with Natural Taurine
1442 MOLECULAR MEDICINE REPORTS 7: 1442-1452, 2013 Transcriptomic and proteomic analysis of human hepatic stellate cells treated with natural taurine JIAN LIANG, XIN DENG, FA‑SHENG WU and YAN‑FANG TANG Ruikang Hospital of Guangxi Traditional Chinese Medical University, Nanning, Guangxi 530011, P.R. China Received September 24, 2012; Accepted February 14, 2013 DOI: 10.3892/mmr.2013.1389 Abstract. The aim of this study was to investigate the Introduction differential expression of genes and proteins between natural taurine (NTau)-treated hepatic stellate cells (HSCs) and Hepatic fibrosis refers to the excessive accumulation of extra- control cells as well as the underlying mechanism of NTau in cellular matrix (ECM) components in the liver. It occurs in inhibiting hepatic fibrosis. A microculture tetrazolium(MTT) most types of chronic liver diseases, including liver cirrhosis, assay was used to analyze the proliferation of NTau‑treated liver failure, and portal hypertension, and often requires liver HSCs. Flow cytometry was performed to compare the transplantation (1). The activation of hepatic stellate cells apoptosis rate between NTau-treated and non-treated HSCs. (HSCs) is an important event by which this cell type, which Proteomic analysis using a combination of 2-dimensional is otherwise quiescent, expresses α-smooth muscle actin gel electrophoresis (2DE) and mass spectrometry (MS) was (α‑SMA), assumes a myofibroblastic phenotype and synthe- conducted to identify the differentially expressed proteins. sizes fibrillar collagens (2,3). Therefore, the inhibition of Microarray analysis was performed to investigate the differ- HSC proliferation, the regulation of the HSC cell cycle, and ential expression of genes and real-time polymerase chain the facilitation of HSC apoptosis are important therapeutic reaction (PCR) was used to validate the results.