Genome-Wide Expression Analysis Suggests Unique Disease-Promoting and Disease-Preventing Signatures in Pemphigus Vulgaris
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Isyte: Integrated Systems Tool for Eye Gene Discovery
Lens iSyTE: Integrated Systems Tool for Eye Gene Discovery Salil A. Lachke,1,2,3,4 Joshua W. K. Ho,1,4,5 Gregory V. Kryukov,1,4,6 Daniel J. O’Connell,1 Anton Aboukhalil,1,7 Martha L. Bulyk,1,8,9 Peter J. Park,1,5,10 and Richard L. Maas1 PURPOSE. To facilitate the identification of genes associated ther investigation. Extension of this approach to other ocular with cataract and other ocular defects, the authors developed tissue components will facilitate eye disease gene discovery. and validated a computational tool termed iSyTE (integrated (Invest Ophthalmol Vis Sci. 2012;53:1617–1627) DOI: Systems Tool for Eye gene discovery; http://bioinformatics. 10.1167/iovs.11-8839 udel.edu/Research/iSyTE). iSyTE uses a mouse embryonic lens gene expression data set as a bioinformatics filter to select candidate genes from human or mouse genomic regions impli- ven with the advent of high-throughput sequencing, the cated in disease and to prioritize them for further mutational Ediscovery of genes associated with congenital birth defects and functional analyses. such as eye defects remains a challenge. We sought to develop METHODS. Microarray gene expression profiles were obtained a straightforward experimental approach that could facilitate for microdissected embryonic mouse lens at three key devel- the identification of candidate genes for developmental disor- opmental time points in the transition from the embryonic day ders, and, as proof-of-principle, we chose defects involving the (E)10.5 stage of lens placode invagination to E12.5 lens primary ocular lens. Opacification of the lens results in cataract, a leading cause of blindness that affects 77 million persons and fiber cell differentiation. -
Primepcr™Assay Validation Report
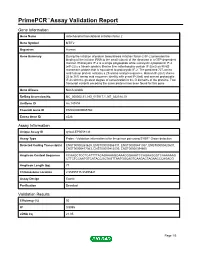
PrimePCR™Assay Validation Report Gene Information Gene Name mitochondrial translational initiation factor 2 Gene Symbol MTIF2 Organism Human Gene Summary During the initiation of protein biosynthesis initiation factor-2 (IF-2) promotes the binding of the initiator tRNA to the small subunit of the ribosome in a GTP-dependent manner. Prokaryotic IF-2 is a single polypeptide while eukaryotic cytoplasmic IF-2 (eIF-2) is a trimeric protein. Bovine liver mitochondria contain IF-2(mt) an 85-kD monomeric protein that is equivalent to prokaryotic IF-2. The predicted 727-amino acid human protein contains a 29-amino acid presequence. Human IF-2(mt) shares 32 to 38% amino acid sequence identity with yeast IF-2(mt) and several prokaryotic IF-2s with the greatest degree of conservation in the G domains of the proteins. Two transcript variants encoding the same protein have been found for this gene. Gene Aliases Not Available RefSeq Accession No. NC_000002.11, NG_017017.1, NT_022184.15 UniGene ID Hs.149894 Ensembl Gene ID ENSG00000085760 Entrez Gene ID 4528 Assay Information Unique Assay ID qHsaCEP0058136 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000263629, ENST00000366137, ENST00000441307, ENST00000420637, ENST00000417363, ENST00000412530, ENST00000394600 Amplicon Context Sequence CCAAGCTCCTCATTTTACAGAAAAGGAAACGGAAATCCAGAAGGGTCAAAAAAG CTTCTCCAATGTCATACCGCTAGTTAATGGCAGTCAAGACTAGAACCCAGACG Amplicon Length (bp) 77 Chromosome Location 2:55495715-55495821 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 95 R2 0.9995 cDNA Cq 21.05 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 79 gDNA Cq 24.56 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

Selective Induction of Apoptosis in HIV-Infected Macrophages
Selective Induction of Apoptosis in HIV-infected Macrophages by Simon Xin Min Dong A thesis submitted in partial fulfillment of the requirements for the Doctorate in Philosophy degree in Microbiology and Immunology Department of Biochemistry, Microbiology, and Immunology Faculty of Medicine University of Ottawa © Simon Xin Min Dong, Ottawa, Canada, 2020 Abstract The eradication of Human Immunodeficiency Virus (HIV) from infected patients is one of the major medical problems of our time, primarily due to HIV reservoir formation. Macrophages play important roles in HIV reservoir formation: once infected, they shield HIV against host anti-viral immune responses and anti-retroviral therapies, help viral spread and establish infection in anatomically protected sites. Thus, it is imperative to selectively induce the apoptosis of HIV-infected macrophages for a complete cure of this disease. I hypothesize that HIV infection dysregulates the expression of some specific genes, which is essential to the survival of infected host cells, and these genes can be targeted to selectively induce the apoptosis of HIV-infected macrophages. My objective is to identify the genes that can be targeted to eradicate HIV reservoir in macrophages, and to briefly elucidate the mechanism of cell death induced by targeting one of the identified genes. A four-step strategy was proposed to reach the goal. First, 90k shRNA lentivirus pool technology and microarray analysis were employed in a genome-wide screen of genes and 28 promising genes were found. Second, siRNA silencing was applied to validate these genes with 2 different HIV-1 viruses; as a result, 4 genes, Cox7a2, Znf484, Cdk2, and Cstf2t, were identified to be novel gene targets. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Evidence for Differential Alternative Splicing in Blood of Young Boys With
Stamova et al. Molecular Autism 2013, 4:30 http://www.molecularautism.com/content/4/1/30 RESEARCH Open Access Evidence for differential alternative splicing in blood of young boys with autism spectrum disorders Boryana S Stamova1,2,5*, Yingfang Tian1,2,4, Christine W Nordahl1,3, Mark D Shen1,3, Sally Rogers1,3, David G Amaral1,3 and Frank R Sharp1,2 Abstract Background: Since RNA expression differences have been reported in autism spectrum disorder (ASD) for blood and brain, and differential alternative splicing (DAS) has been reported in ASD brains, we determined if there was DAS in blood mRNA of ASD subjects compared to typically developing (TD) controls, as well as in ASD subgroups related to cerebral volume. Methods: RNA from blood was processed on whole genome exon arrays for 2-4–year-old ASD and TD boys. An ANCOVA with age and batch as covariates was used to predict DAS for ALL ASD (n=30), ASD with normal total cerebral volumes (NTCV), and ASD with large total cerebral volumes (LTCV) compared to TD controls (n=20). Results: A total of 53 genes were predicted to have DAS for ALL ASD versus TD, 169 genes for ASD_NTCV versus TD, 1 gene for ASD_LTCV versus TD, and 27 genes for ASD_LTCV versus ASD_NTCV. These differences were significant at P <0.05 after false discovery rate corrections for multiple comparisons (FDR <5% false positives). A number of the genes predicted to have DAS in ASD are known to regulate DAS (SFPQ, SRPK1, SRSF11, SRSF2IP, FUS, LSM14A). In addition, a number of genes with predicted DAS are involved in pathways implicated in previous ASD studies, such as ROS monocyte/macrophage, Natural Killer Cell, mTOR, and NGF signaling. -

Dynamics of the Linc Complex
DYNAMICS OF THE LINC COMPLEX Loic Gazquez University of Manchester School of Biological Sciences 2017 A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Biology, Medicine and Health TABLE OF CONTENTS Table of Contents .................................................................................................................................... 2 Abstract.................................................................................................................................................... 4 Declaration .............................................................................................................................................. 5 Copyright statement ................................................................................................................................ 5 Acknowledgements ................................................................................................................................. 6 I. Introduction .................................................................................................................................. 7 Nuclear Envelope and the LINC complex ................................................................................... 7 I. 1.1. The first LINC component: the SUN ................................................................................ 8 I. 1.2. The second LINC component: the KASH ........................................................................ 8 I. 1.1. Structure -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g. -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest. -

Role of Phytochemicals in Colon Cancer Prevention: a Nutrigenomics Approach
Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan J van Erk Promotor: Prof. Dr. P.J. van Bladeren Hoogleraar in de Toxicokinetiek en Biotransformatie Wageningen Universiteit Co-promotoren: Dr. Ir. J.M.M.J.G. Aarts Universitair Docent, Sectie Toxicologie Wageningen Universiteit Dr. Ir. B. van Ommen Senior Research Fellow Nutritional Systems Biology TNO Voeding, Zeist Promotiecommissie: Prof. Dr. P. Dolara University of Florence, Italy Prof. Dr. J.A.M. Leunissen Wageningen Universiteit Prof. Dr. J.C. Mathers University of Newcastle, United Kingdom Prof. Dr. M. Müller Wageningen Universiteit Dit onderzoek is uitgevoerd binnen de onderzoekschool VLAG Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Marjan Jolanda van Erk Proefschrift ter verkrijging van graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof.Dr.Ir. L. Speelman, in het openbaar te verdedigen op vrijdag 1 oktober 2004 des namiddags te vier uur in de Aula Title Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Author Marjan Jolanda van Erk Thesis Wageningen University, Wageningen, the Netherlands (2004) with abstract, with references, with summary in Dutch ISBN 90-8504-085-X ABSTRACT Role of phytochemicals in colon cancer prevention: a nutrigenomics approach Specific food compounds, especially from fruits and vegetables, may protect against development of colon cancer. In this thesis effects and mechanisms of various phytochemicals in relation to colon cancer prevention were studied through application of large-scale gene expression profiling. Expression measurement of thousands of genes can yield a more complete and in-depth insight into the mode of action of the compounds. -
Integrated Bioinformatics Analysis of Aberrantly-Methylated
Shen et al. BMC Ophthalmology (2020) 20:119 https://doi.org/10.1186/s12886-020-01392-2 RESEARCH ARTICLE Open Access Integrated bioinformatics analysis of aberrantly-methylated differentially- expressed genes and pathways in age- related macular degeneration Yinchen Shen1,2†,MoLi3†, Kun Liu1,2, Xiaoyin Xu1,2, Shaopin Zhu1,2, Ning Wang1,2, Wenke Guo4, Qianqian Zhao4, Ping Lu4, Fudong Yu4 and Xun Xu1,2* Abstract Background: Age-related macular degeneration (AMD) represents the leading cause of visual impairment in the aging population. The goal of this study was to identify aberrantly-methylated, differentially-expressed genes (MDEGs) in AMD and explore the involved pathways via integrated bioinformatics analysis. Methods: Data from expression profile GSE29801 and methylation profile GSE102952 were obtained from the Gene Expression Omnibus database. We analyzed differentially-methylated genes and differentially-expressed genes using R software. Functional enrichment and protein–protein interaction (PPI) network analysis were performed using the R package and Search Tool for the Retrieval of Interacting Genes online database. Hub genes were identified using Cytoscape. Results: In total, 827 and 592 genes showed high and low expression, respectively, in GSE29801; 4117 hyper-methylated genes and 511 hypo-methylated genes were detected in GSE102952. Based on overlap, we categorized 153 genes as hyper-methylated, low-expression genes (Hyper-LGs) and 24 genes as hypo-methylated, high-expression genes (Hypo-HGs). Four Hyper-LGs (CKB, PPP3CA, TGFB2, SOCS2) overlapped with AMD risk genes in the Public Health Genomics and Precision Health Knowledge Base. KEGG pathway enrichment analysis indicated that Hypo-HGs were enriched in the calcium signaling pathway, whereas Hyper-LGs were enriched in sphingolipid metabolism. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

1 Identification of Marbling Gene Loci in Commercial Pigs in Canadian Herds
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 21 June 2018 doi:10.20944/preprints201806.0330.v1 Peer-reviewed version available at Agriculture 2018, 8, 122; doi:10.3390/agriculture8080122 1 Identification of marbling gene loci in commercial pigs in Canadian herds. 2 W. J. Meadus, P. Duff, J. Roberts, J.L. Zantinge, I. Larsen, J.L. Aalhus, M. Juraez. 3 Agri-Food & Agriculture Canada, Lacombe Research Centre, 6000 C&E Trail, Lacombe, Alberta, Canada. T4L 1W1. 4 5 Abstract. 6 We examined the amount of marbling and tested the genome of boars from 5 breeds of Duroc, Iberian, 7 Lacombe, Berkshire and Pietrian that were commercially available for a swine herd in Canada. The 8 marbling was ranked according to the amount of intramuscular fat % obtained in loin chops consisting of 9 the longissimus dorsi muscle. The genetics were analysed by genome wide association study using 10 80,000 single nuclear polymorphism (SNP) microarrays. Our samples had pork that achieved > 7 % IMF 11 from 110kg animals. Meta-analysis revealed SNP markers that were associated with the highest marbled 12 pork chops on chromosomes 5, 7, and 16. Using the susScr 11.1 map, we determined that the nearest 13 genes were SSNP, Rh glycoprotein and EGFLAM. We tested a sub-population of Duroc sired animals and 14 found a different set of markers close to GRLB and KCNJ3 on chromosomes 8 and 15. Based on our 15 sample, we can achieve pork with good marbling from animals conventionally raised to standard market 16 weights of 110kg. The choice of a good marbling line of pig is not necessarily breed specific.