Genome-Wide Analysis of Epigenetic Silencing Identifies BEX1 and BEX2 As Candidate Tumor Suppressor Genes in Malignant Glioma
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Information to Mammadova-Bach Et Al., “Laminin Α1 Orchestrates VEGFA Functions in the Ecosystem of Colorectal Carcinogenesis”
Supplemental information to Mammadova-Bach et al., “Laminin α1 orchestrates VEGFA functions in the ecosystem of colorectal carcinogenesis” Supplemental material and methods Cloning of the villin-LMα1 vector The plasmid pBS-villin-promoter containing the 3.5 Kb of the murine villin promoter, the first non coding exon, 5.5 kb of the first intron and 15 nucleotides of the second villin exon, was generated by S. Robine (Institut Curie, Paris, France). The EcoRI site in the multi cloning site was destroyed by fill in ligation with T4 polymerase according to the manufacturer`s instructions (New England Biolabs, Ozyme, Saint Quentin en Yvelines, France). Site directed mutagenesis (GeneEditor in vitro Site-Directed Mutagenesis system, Promega, Charbonnières-les-Bains, France) was then used to introduce a BsiWI site before the start codon of the villin coding sequence using the 5’ phosphorylated primer: 5’CCTTCTCCTCTAGGCTCGCGTACGATGACGTCGGACTTGCGG3’. A double strand annealed oligonucleotide, 5’GGCCGGACGCGTGAATTCGTCGACGC3’ and 5’GGCCGCGTCGACGAATTCACGC GTCC3’ containing restriction site for MluI, EcoRI and SalI were inserted in the NotI site (present in the multi cloning site), generating the plasmid pBS-villin-promoter-MES. The SV40 polyA region of the pEGFP plasmid (Clontech, Ozyme, Saint Quentin Yvelines, France) was amplified by PCR using primers 5’GGCGCCTCTAGATCATAATCAGCCATA3’ and 5’GGCGCCCTTAAGATACATTGATGAGTT3’ before subcloning into the pGEMTeasy vector (Promega, Charbonnières-les-Bains, France). After EcoRI digestion, the SV40 polyA fragment was purified with the NucleoSpin Extract II kit (Machery-Nagel, Hoerdt, France) and then subcloned into the EcoRI site of the plasmid pBS-villin-promoter-MES. Site directed mutagenesis was used to introduce a BsiWI site (5’ phosphorylated AGCGCAGGGAGCGGCGGCCGTACGATGCGCGGCAGCGGCACG3’) before the initiation codon and a MluI site (5’ phosphorylated 1 CCCGGGCCTGAGCCCTAAACGCGTGCCAGCCTCTGCCCTTGG3’) after the stop codon in the full length cDNA coding for the mouse LMα1 in the pCIS vector (kindly provided by P. -

FKBP52 Regulates TRPC3-Dependent Ca<Sup>
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs231506. doi:10.1242/jcs.231506 RESEARCH ARTICLE FKBP52 regulates TRPC3-dependent Ca2+ signals and the hypertrophic growth of cardiomyocyte cultures Sandra Bandleon1, Patrick P. Strunz1, Simone Pickel2, Oleksandra Tiapko3, Antonella Cellini1, Erick Miranda-Laferte2 and Petra Eder-Negrin1,* ABSTRACT A single TRPC subunit is composed of six transmembrane The transient receptor potential (TRP; C-classical, TRPC) channel domains with a pore-forming loop connecting the transmembrane ‘ ’ TRPC3 allows a cation (Na+/Ca2+) influx that is favored by the domains 5 and 6, a preserved 25 amino acid sequence called a TRP domain and two cytosolic domains, an N-terminal ankyrin repeat stimulation of Gq protein-coupled receptors (GPCRs). An enhanced TRPC3 activity is related to adverse effects, including pathological domain and a C-terminal coiled-coil domain (Eder et al., 2007; Fan hypertrophy in chronic cardiac disease states. In the present study, et al., 2018). The cytosolic domains mediate ion channel formation we identified FK506-binding protein 52 (FKBP52, also known as and are implicated in ion channel regulation and plasma membrane FKBP4) as a novel interaction partner of TRPC3 in the heart. FKBP52 targeting (Eder et al., 2007). Among several protein interaction sites, was recovered from a cardiac cDNA library by a C-terminal TRPC3 the C-terminus of all TRPC subunits harbors a highly conserved fragment (amino acids 742–848) in a yeast two-hybrid screen. proline-rich sequence that corresponds to the binding domain in the Drosophila Downregulation of FKBP52 promoted a TRPC3-dependent photoreceptor channel TRPL for the FK506-binding hypertrophic response in neonatal rat cardiomyocytes (NRCs). -
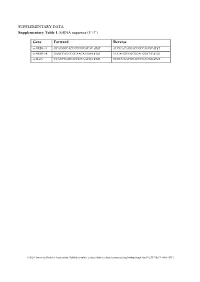
Supplementary Tables and Figures
SUPPLEMENTARY DATA Supplementary Table 1. SiRNA sequence (5’-3’) Gene Forward Reverse si-HRD1-1# GCAUGGCAGUCCUGUACAU dTdT AUGUACAGGACUGCCAUGC dTdT si-HRD1-2# GAGCCAUCCGCAACAUGAA dTdT UUCAUGUUGCGGAUGGCUC dTdT si-MafA CCAUCGAGUACGUCAACGA dTdT UCGUUGACGUACUCGAUGG dTdT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR (5’-3’) Gene Forward Reverse human HRD1 GCTCACGCCTACTACCTCAAA GCCAGACAAGTCTCTGTGACG mouse mafA AAGCGGCGCACGCTCAAGAA GGTCCCGCTCCTTGGCCAGA mouse insulin1 CACTTCCTACCCCTGCTGG ACCACAAAGATGCTGTTTGACA mouse β-actin AGGCCAACCGTGAAAAGATG AGAGCATAGCCCTCGTAGATGG human β-actin CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 3. Primer sequences for ChIP (5’-3’) Gene promoter Forward Reverse mouse Insulin1, 2 GGAACTGTGAAACAGTCCAAGG CCCCCTGGACTTTGCTGTTTG ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 4. Primer sequences for PCR (5’-3’) Gene Forward Reverse HRD1-pDsred CCCAAGCTTATGTTCCGCACCGCAGT GGGGTACCCAGTGGGCAACAGGGG HRD1-pCMV- Flag GGGGTACCATGTTCCGCACCGCAGT CCCAAGCTTGTGGGCAACAGGGGACT C HRD1-pCMV-HA GGCCATGGGCCATATGGGATCCTTCC AGGGATGCCACCCGGGGATCCTCAGT GCACCGCAGTGATG GGGCAACAGGGGAC HRD1-N-HA GGCCATGGGCCATATGGGATCCTTCC -

Senescence Inhibits the Chaperone Response to Thermal Stress
SUPPLEMENTAL INFORMATION Senescence inhibits the chaperone response to thermal stress Jack Llewellyn1, 2, Venkatesh Mallikarjun1, 2, 3, Ellen Appleton1, 2, Maria Osipova1, 2, Hamish TJ Gilbert1, 2, Stephen M Richardson2, Simon J Hubbard4, 5 and Joe Swift1, 2, 5 (1) Wellcome Centre for Cell-Matrix Research, Oxford Road, Manchester, M13 9PT, UK. (2) Division of Cell Matrix Biology and Regenerative Medicine, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, M13 9PL, UK. (3) Current address: Department of Biomedical Engineering, University of Virginia, Box 800759, Health System, Charlottesville, VA, 22903, USA. (4) Division of Evolution and Genomic Sciences, School of Biological Sciences, Faculty of Biology, Medicine and Health, Manchester Academic Health Science Centre, University of Manchester, Manchester, M13 9PL, UK. (5) Correspondence to SJH ([email protected]) or JS ([email protected]). Page 1 of 11 Supplemental Information: Llewellyn et al. Chaperone stress response in senescence CONTENTS Supplemental figures S1 – S5 … … … … … … … … 3 Supplemental table S6 … … … … … … … … 10 Supplemental references … … … … … … … … 11 Page 2 of 11 Supplemental Information: Llewellyn et al. Chaperone stress response in senescence SUPPLEMENTAL FIGURES Figure S1. A EP (passage 3) LP (passage 16) 200 µm 200 µm 1.5 3 B Mass spectrometry proteomics (n = 4) C mRNA (n = 4) D 100k EP 1.0 2 p < 0.0001 p < 0.0001 LP p < 0.0001 p < 0.0001 ) 0.5 1 2 p < 0.0001 p < 0.0001 10k 0.0 0 -0.5 -1 Cell area (µm Cell area fold change vs. EP fold change vs. -

FK506-Binding Protein 12.6/1B, a Negative Regulator of [Ca<Sup>2+
University of Kentucky UKnowledge Pharmacology and Nutritional Sciences Faculty Pharmacology and Nutritional Sciences Publications Repository Citation Gant, John C.; Blalock, Eric M.; Chen, Kuey-Chu; Kadish, Inga; Thibault, Olivier; Porter, Nada M.; and Landfield, Philip W., "FK506-Binding Protein 12.6/1b, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats" (2018). Pharmacology and Nutritional Sciences Faculty Publications. 65. https://uknowledge.uky.edu/pharmacol_facpub/65 This Article is brought to you for free and open access by the Pharmacology and Nutritional Sciences at UKnowledge. It has been accepted for inclusion in Pharmacology and Nutritional Sciences Faculty Publications by an authorized administrator of UKnowledge. For more information, please contact [email protected]. 1-24-2018 FK506-Binding Protein 12.6/1b, a Negative Regulator of [Ca2+], Rescues Memory and Restores Genomic Regulation in the Hippocampus of Aging Rats John C. Gant University of Kentucky, [email protected] Eric M. Blalock University of Kentucky, [email protected] Kuey-Chu Chen University of Kentucky, [email protected] Inga Kadish University of Alabama at Birmingham Olivier Thibault University of Kentucky, [email protected] See next page for additional authors Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/pharmacol_facpub Part of the Cell and Developmental Biology Commons, Diseases Commons, Medical Nutrition Commons, Medical Pharmacology Commons, Neuroscience and Neurobiology Commons, and the Pharmacology, Toxicology and Environmental Health Commons Authors John C. Gant, Eric M. -

Homologs of Human Dengue-Resistance Genes, FKBP1B and ATCAY, Confer Antiviral Resistance in Aedes Aegypti Mosquitoes
insects Article Homologs of Human Dengue-Resistance Genes, FKBP1B and ATCAY, Confer Antiviral Resistance in Aedes aegypti Mosquitoes Seokyoung Kang 1,*,†, Dongyoung Shin 2,†, Derrick K. Mathias 2, Berlin Londono-Renteria 3 , Mi Young Noh 4, Tonya M. Colpitts 5, Rhoel R. Dinglasan 1, Yeon Soo Han 4 and Young S. Hong 6,* 1 Emerging Pathogens Institute and the Department of Infectious Diseases and Immunology, College of Veterinary Medicine, University of Florida, Gainesville, FL 32611, USA; [email protected]fl.edu 2 Florida Medical Entomology Laboratory, Department of Nematology and Entomology, Institute of Food and Agricultural Sciences, University of Florida, Vero Beach, FL 32962, USA; dshin@ufl.edu (D.S.); d.mathias@ufl.edu (D.K.M.) 3 Department of Entomology, College of Agriculture, Kansas State University, Manhattan, KS 66506, USA; [email protected] 4 Department of Agricultural Biology, College of Agriculture and Life Sciences, Chonnam National University, Gwang-ju 500-757, Korea; [email protected] (M.Y.N.); [email protected] (Y.S.H.) 5 Department of Microbiology, Boston University School of Medicine, National Emerging Infectious Diseases Laboratories, Boston, MA 02118, USA; [email protected] 6 Access Bio, Inc., Somerset, NJ 08873, USA * Correspondence: skang1@ufl.edu (S.K.); [email protected] (Y.S.H.) † These authors contributed equally to this work. Received: 1 September 2018; Accepted: 29 January 2019; Published: 2 February 2019 Abstract: Dengue virus (DENV) is transmitted by mosquitoes and is a major public health concern. The study of innate mosquito defense mechanisms against DENV have revealed crucial roles for the Toll, Imd, JAK-STAT, and RNAi pathways in mediating DENV in the mosquito. -

Inhibition of the FKBP Family of Peptidyl Prolyl Isomerases Induces Abortive Translocation and Degradation of the Cellular Prion Protein
Inhibition of the FKBP Family of Peptidyl Prolyl Isomerases Induces Abortive Translocation and Degradation of the Cellular Prion Protein by Maxime Sawicki A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Biochemistry University of Toronto © Copyright by Maxime Sawicki 2015 Inhibition of the FKBP Family of Peptidyl Prolyl Isomerases Induces Abortive Translocation and Degradation of the Cellular Prion Protein Maxime Sawicki Master of Science Department of Biochemistry University of Toronto 2015 Abstract Prion disorders are a class of neurodegenerative diseases that feature a structural change of the prion protein from its cellular form (PrPC) into its scrapie form (PrPSc). As these disorders are currently incurable, there is a crucial need for novel therapeutic agents. Here, the FDA-approved immunosuppressive drug FK506 was shown to cause an attenuation in the endoplasmic reticulum (ER) translocation of PrPC by exacerbating an intrinsic inefficiency of PrP’s ER-targeting signal sequence, effectively causing the proteasomal degradation of PrPC. Furthermore, the depletion of FKBP10 also caused the degradation of PrPC but at a later stage following translocation into the ER. Additionally, novel FK506 analogues with reduced immunosuppressive properties were shown to be as efficacious as FK506 in downregulating PrPC. Finally, both FK506 treatment and FKBP10 depletion were shown to reduce the levels of PrPSc in chronically infected cell models. These findings offer a new insight into the development of treatments against prion disorders. ii Acknowledgments The completion of the present thesis would not have been possible without the help and support of a number of people. First and foremost, I would like to thank my supervisor, Dr David Williams, for his constant guidance and expertise that allowed me to successfully complete my degree, as well as my committee members, Dr John Glover and Dr Gerold Schmitt-Ulms, for their invaluable advice and suggestions over the course of this project. -

Downloaded and the Kinasefrom Domainthe Pubmed of TOR Server As Input at the Templates
biomolecules Review Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities Andrzej Galat Service d’Ingénierie Moléculaire des Protéines (SIMOPRO), CEA, Université Paris-Saclay, F-91191 Gif/Yvette, France; [email protected]; Fax: +33-169089137 Academic Editor: John A. Carver Received: 24 August 2017; Accepted: 26 September 2017; Published: 29 September 2017 Abstract: Analyses of sequences and structures of the cyclosporine A (CsA)-binding proteins (cyclophilins) and the immunosuppressive macrolide FK506-binding proteins (FKBPs) have revealed that they exhibit peculiar spatial distributions of charges, their overall hydrophobicity indexes vary within a considerable level whereas their points isoelectric (pIs) are contained from 4 to 11. These two families of peptidylprolyl cis/trans isomerases (PPIases) have several distinct functional attributes such as: (1) high affinity binding to some pharmacologically-useful hydrophobic macrocyclic drugs; (2) diversified binding epitopes to proteins that may induce transient manifolds with altered flexibility and functional fitness; and (3) electrostatic interactions between positively charged segments of PPIases and negatively charged intracellular entities that support their spatial integration. These three attributes enhance binding of PPIase/pharmacophore complexes to diverse intracellular entities, some of which perturb signalization pathways causing immunosuppression and other system-altering phenomena in humans. Keywords: PPIase; FKBP; cyclophilin; rapamycin; FK506 1. Introduction About forty years ago, two different types of macrocyclic molecules were isolated and shown to possess immunosuppressive activities, such as the cyclic peptide containing non-standard amino acid residues (AAs) cyclosporine A (CsA) and its homologues [1], and two polyketides having L-pipecolic acid ring, namely immunosuppressive macrolide FK506 [2] and rapamycin [3,4]. -

Supplementary Materials (PDF)
Proteomics of the mediodorsal thalamic nucleus in gastric ulcer induced by restraint-water-immersion-stress Sheng-Nan Gong, Jian-Ping Zhu, Ying-Jie Ma, Dong-Qin Zhao Table S1. The entire list of 2,853 proteins identified between the control and stressed groups Protein NO Protein name Gene name Accession No LogRatio 1 Tubulin alpha-1A chain Tuba1a TBA1A_RAT 0.2320 2 Spectrin alpha chain, non-erythrocytic 1 Sptan1 A0A0G2JZ69_RAT -0.0291 3 ATP synthase subunit alpha, mitochondrial Atp5f1a ATPA_RAT -0.1155 4 Tubulin beta-2B chain Tubb2b TBB2B_RAT 0.0072 5 Actin, cytoplasmic 2 Actg1 ACTG_RAT 0.0001 Sodium/potassium-transporting ATPase Atp1a2 6 subunit alpha-2 AT1A2_RAT -0.0716 7 Spectrin beta chain Sptbn1 A0A0G2K8W9_RAT -0.1158 8 Clathrin heavy chain 1 Cltc CLH1_RAT 0.0788 9 Dihydropyrimidinase-related protein 2 Dpysl2 DPYL2_RAT -0.0696 10 Glyceraldehyde-3-phosphate dehydrogenase Gapdh G3P_RAT -0.0687 Sodium/potassium-transporting ATPase Atp1a3 11 subunit alpha-3 AT1A3_RAT 0.0391 12 ATP synthase subunit beta, mitochondrial Atp5f1b ATPB_RAT 0.1772 13 Cytoplasmic dynein 1 heavy chain 1 Dync1h1 M0R9X8_RAT 0.0527 14 Myelin basic protein transcript variant N Mbp I7EFB0_RAT 0.0696 15 Microtubule-associated protein Map2 F1LNK0_RAT -0.1053 16 Pyruvate kinase PKM Pkm KPYM_RAT -0.2608 17 D3ZQQ5_RAT 0.0087 18 Plectin Plec F7F9U6_RAT -0.0076 19 14-3-3 protein zeta/delta Ywhaz A0A0G2JV65_RAT -0.2431 20 2',3'-cyclic-nucleotide 3'-phosphodiesterase Cnp CN37_RAT -0.0495 21 Creatine kinase B-type Ckb KCRB_RAT -0.0514 Voltage-dependent anion-selective channel -

Oxidized Phospholipids Regulate Amino Acid Metabolism Through MTHFD2 to Facilitate Nucleotide Release in Endothelial Cells
ARTICLE DOI: 10.1038/s41467-018-04602-0 OPEN Oxidized phospholipids regulate amino acid metabolism through MTHFD2 to facilitate nucleotide release in endothelial cells Juliane Hitzel1,2, Eunjee Lee3,4, Yi Zhang 3,5,Sofia Iris Bibli2,6, Xiaogang Li7, Sven Zukunft 2,6, Beatrice Pflüger1,2, Jiong Hu2,6, Christoph Schürmann1,2, Andrea Estefania Vasconez1,2, James A. Oo1,2, Adelheid Kratzer8,9, Sandeep Kumar 10, Flávia Rezende1,2, Ivana Josipovic1,2, Dominique Thomas11, Hector Giral8,9, Yannick Schreiber12, Gerd Geisslinger11,12, Christian Fork1,2, Xia Yang13, Fragiska Sigala14, Casey E. Romanoski15, Jens Kroll7, Hanjoong Jo 10, Ulf Landmesser8,9,16, Aldons J. Lusis17, 1234567890():,; Dmitry Namgaladze18, Ingrid Fleming2,6, Matthias S. Leisegang1,2, Jun Zhu 3,4 & Ralf P. Brandes1,2 Oxidized phospholipids (oxPAPC) induce endothelial dysfunction and atherosclerosis. Here we show that oxPAPC induce a gene network regulating serine-glycine metabolism with the mitochondrial methylenetetrahydrofolate dehydrogenase/cyclohydrolase (MTHFD2) as a cau- sal regulator using integrative network modeling and Bayesian network analysis in human aortic endothelial cells. The cluster is activated in human plaque material and by atherogenic lipo- proteins isolated from plasma of patients with coronary artery disease (CAD). Single nucleotide polymorphisms (SNPs) within the MTHFD2-controlled cluster associate with CAD. The MTHFD2-controlled cluster redirects metabolism to glycine synthesis to replenish purine nucleotides. Since endothelial cells secrete purines in response to oxPAPC, the MTHFD2- controlled response maintains endothelial ATP. Accordingly, MTHFD2-dependent glycine synthesis is a prerequisite for angiogenesis. Thus, we propose that endothelial cells undergo MTHFD2-mediated reprogramming toward serine-glycine and mitochondrial one-carbon metabolism to compensate for the loss of ATP in response to oxPAPC during atherosclerosis. -

The Human Fk506binding Proteins: Characterization of Human FKBP19
The human FK506-binding proteins: characterization of human FKBP19 Article (Accepted Version) Rulten, Stuart L, Kinloch, Ross A, Tateossian, Hilda, Robinson, Colin, Gettins, Lucy and Kay, John E (2006) The human FK506-binding proteins: characterization of human FKBP19. Mammalian Genome, 17 (4). pp. 322-331. ISSN 0938-8990 This version is available from Sussex Research Online: http://sro.sussex.ac.uk/id/eprint/22944/ This document is made available in accordance with publisher policies and may differ from the published version or from the version of record. If you wish to cite this item you are advised to consult the publisher’s version. Please see the URL above for details on accessing the published version. Copyright and reuse: Sussex Research Online is a digital repository of the research output of the University. Copyright and all moral rights to the version of the paper presented here belong to the individual author(s) and/or other copyright owners. To the extent reasonable and practicable, the material made available in SRO has been checked for eligibility before being made available. Copies of full text items generally can be reproduced, displayed or performed and given to third parties in any format or medium for personal research or study, educational, or not-for-profit purposes without prior permission or charge, provided that the authors, title and full bibliographic details are credited, a hyperlink and/or URL is given for the original metadata page and the content is not changed in any way. http://sro.sussex.ac.uk The Human FK506-Binding Proteins: Characterisation of Human FKBP19 Stuart L Rulten1*, Ross A Kinloch2, Hilda Tateossian3, Colin Robinson2, Lucy Gettins2 and John E Kay4. -

First Line of Title
THE ABDOMINAL FAT CONTRIBUTION TO ADIPOSITY IN CHICKENS DIVERGENTLY SELECTED FOR FATNESS OR GROWTH: CROSS-MODEL ELUCIDATION AND VALIDATION OF GENE EXPRESSION by Christopher William Resnyk A thesis submitted to the Faculty of the University of Delaware in partial fulfillment of the requirements for the degree of Master of Science in Animal Science Summer 2013 © 2013 Christopher William Resnyk All Rights Reserved THE ABDOMINAL FAT CONTRIBUTION TO ADIPOSITY IN CHICKENS DIVERGENTLY SELECTED FOR FATNESS OR GROWTH: CROSS-MODEL ELUCIDATION AND VALIDATION OF GENE EXPRESSION by Christopher William Resnyk Approved: __________________________________________________________ Larry A. Cogburn III, Ph.D. Professor in charge of thesis on behalf of the Advisory Committee Approved: __________________________________________________________ Jack Gelb, Jr., Ph.D. Chair of the Department of Animal and Food Sciences Approved: __________________________________________________________ Mark Rieger, Ph.D. Dean of the College of Agriculture and Natural Resources Approved: __________________________________________________________ James G. Richards, Ph.D. Vice Provost for Graduate and Professional Education ACKNOWLEDGMENTS This work was supported by a grant from the United States Department of Agriculture, Initiative for Future Agricultural and Food Systems, Animal Genome Program (USDA-IFAFS; Award # 00-52100-9614) and a grant from the USDA Cooperative State Research, Education, and Extension Service (CSREES; Award # 2009-34562-20008) to the Avian Biosciences Center, University of Delaware. I want to thank my collaborators on this work: Sam Aggrey, Wilfrid Carre, Chuming Chen, Larry Cogburn, Michel Duclos, Elisabeth Le Bihan-Duval, Hongzhan Huang, Tom Porter, Jean Simon, Xiaofei Wang, and Cathy Wu. Further, the longitudinal study and tissue sampling of the divergent lines required the contribution of a large number of scientists and technicians from the laboratories of INRA UR83 and from the breeding facilities in Nouzilly, France.