Non-Genomic Action of Steroid Hormones: More Questions Than Answers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Curriculum Vitae
Professor Barry V L Potter Publications (1979 - 2015) Papers in refereed journals, patents, reviews and book chapters JOURNAL PAPERS 1. The effect of 17O and the magnitude of the 18O isotope shift in 31P nuclear magnetic resonance spectroscopy. G Lowe, B V L Potter, B S Sproat and W E Hull, J Chem Soc Chem Commun (1979) 733- 735. 2. Bacteriostatic properties of fluoro-analogues of 5(2-hydroxyethyl)-4-methyl thiazole, a metabolic intermediate in thiamine biosynthesis. G Lowe and B V L Potter, J Chem Soc Perkin Trans I (1980) 9, 2026-2028. 3. The synthesis, absolute configuration and circular dichroism of the enantiomers of fluorosuccinic acid. G Lowe and B V L Potter, J Chem Soc Perkin Trans I (1980) 9, 2029-2032. 4. Evidence against a step-wise mechanism for the fumarase-catalysed dehydration of (2S)- malate. V T Jones, G Lowe and B V L Potter, Eur J Biochem (1980) 108, 433-437. 5. A stereochemical investigation of the cyclisation of D-glucose-6[16O,17O,18O]-phosphate and adenosine 5'[16O,17O,18O]-phosphate. R L Jarvest, G Lowe and B V L Potter, J Chem Soc Chem Commun (1980) 1142-1145. 6. The stereochemistry of phosphoryl transfer. G Lowe, P M Cullis, R L Jarvest, B V L Potter and B S Sproat, Phil Trans Roy Soc Lond Ser B (1981) 293, 75-92. 7. The stereochemistry of 2-substituted-2-oxo-4,5-diphenyl-1,3,2-dioxaphospholanes and the related chiral [16O,17O,18O]-phosphate monoesters. P M Cullis, R L Jarvest, G Lowe and B V L Potter, J Chem Soc Chem Commun (1981) 245- 246. -

Version Introducing VHIO Foreword
2017SCIENTIFIC REPORT Vall d'Hebron Institute of Oncology (VHIO) CELLEX CENTER C/Natzaret, 115 – 117 08035 Barcelona, Spain Tel. +34 93 254 34 50 www.vhio.net Direction: Amanda Wren Design: Parra estudio Photography: Katherin Wermke Vall d'Hebron Institute of Oncology (VHIO) | 2018 VHIO Scientific Report 2017 Index Scientific Report INTRODUCING VHIO CORE TECHNOLOGIES 02 Foreword 92 Cancer Genomics Group 08 2017: marking the next chapter of 94 Molecular Oncology Group VHIO's translational story 96 Proteomics Group 18 Scientific Productivity: research articles VHIO'S TRANSVERSAL CLINICAL 19 Selection of some of the most relevant TRIALS CORE SERVICES & UNITS articles by VHIO researchers published in 2017 100 Clinical Trials Office 25 A Golden Decade: reflecting on the 102 Research Unit for Molecular Therapy past 10 years of VHIO's translational of Cancer (UITM) – ”la Caixa” success story 104 Clinical Research Oncology Nurses 106 Clinical Research Oncology Pharmacy PRECLINICAL RESEARCH Unit 49 From the Director 50 Experimental Therapeutics Group RECENTLY INCORPORATED GROUPS 52 Growth Factors Group 110 Cellular Plasticity & Cancer Group 54 Mouse Models of Cancer Therapies 112 Experimental Hematology Group Group 114 Tumor Immunology 56 Tumor Biomarkers Group & Immunotherapy Group 116 Applied Genetics of Metastatic Cancer TRANSLATIONAL RESEARCH Group 61 From the Director 118 Chromatin Dynamics in Cancer Group 62 Gene Expression & Cancer Group 120 Prostate Cancer Translational Research Group 64 Stem Cells & Cancer Group 122 Radiomics Group CLINICAL -
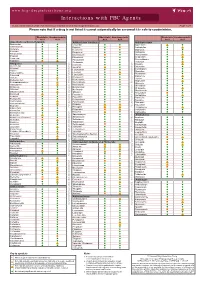
Interactions with PBC Agents
www.hep-druginteractions.org Interactions with PBC Agents Charts created March 2020. Full information available at www.hep-druginteractions.org Page 1 of 4 Please note that if a drug is not listed it cannot automatically be assumed it is safe to coadminister. Obeticholic Ursodeoxycholic Obeticholic Ursodeoxycholic Obeticholic Ursodeoxycholic Acid Acid Acid Acid Acid Acid Anaesthetics & Muscle Relaxants Antibacterials (continued) Antidepressants Bupivacaine Cloxacillin Agomelatine Cisatracurium Dapsone Amitriptyline Isoflurane Delamanid Bupropion Ketamine Ertapenem Citalopram Nitrous oxide Erythromycin Clomipramine Propofol Ethambutol Desipramine Thiopental Flucloxacillin Desvenlafaxine Tizanidine Gentamicin Dosulepin Analgesics Imipenem Doxepin Aceclofenac Isoniazid Duloxetine Alfentanil Escitalopram Aspirin Levofloxacin Linezolid Fluoxetine Buprenorphine Fluvoxamine Lymecycline distribution. Celecoxib Imipramine Meropenem Codeine Lithium Methenamine Dexketoprofen Maprotiline Metronidazole Dextropropoxyphene Mianserin Moxifloxacin Diamorphine Milnacipran Diclofenac Nitrofurantoin only. Not for distribution. for only. Not Mirtazapine Diflunisal Norfloxacin Moclobemide Dihydrocodeine Ofloxacin Nefazodone Etoricoxib Penicillin V Nortriptyline Fentanyl Piperacillin Paroxetine Flurbiprofen Pivmecillinam Sertraline Hydrocodone use ersonal Pyrazinamide Tianeptine Hydromorphone Rifabutin Trazodone Ibuprofen Rifampicin -

(12) United States Patent (10) Patent No.: US 9.498,431 B2 Xu Et Al
USOO9498431B2 (12) United States Patent (10) Patent No.: US 9.498,431 B2 Xu et al. (45) Date of Patent: Nov. 22, 2016 (54) CONTROLLED RELEASING COMPOSITION 7,053,134 B2 * 5/2006 Baldwin et al. .............. 522,154 2004/0058056 A1 3/2004 Osaki et al. ................... 427.2.1 (76) Inventors: Jianjian Xu, Hefei (CN); Shiliang 2005/0037047 A1 2/2005 Song Wang, Hefei (CN); Manzhi Ding 2007/0055364 A1* 3/2007 Hossainy .................. A61F 2/82 s: s s 623, 1.38 Hefei (CN) 2008/0274194 A1* 11/2008 Miller .................... A61K 9.146 424/489 (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 FOREIGN PATENT DOCUMENTS U.S.C. 154(b) by 0 days. CN 1208.610 A 2, 1999 (21) Appl. No.: 13/133,656 EP O251680 A2 1, 1988 JP S63-22516. A 1, 1988 JP H1O-310518 A 11, 1998 (22) PCT Filed: Dec. 10, 2009 WO 96,10395 A1 4f1996 WO WO 2005.000277 A1 * 1, 2005 (86). PCT No.: PCT/CN2009/075468 WO 2007 115045 A2 10, 2007 WO 2008/OO2657 A2 1, 2008 S 371 (c)(1), WO 2008OO2657 A2 1, 2008 (2), (4) Date: Jun. 9, 2011 WO 2008041246 A2 4/2008 (87) PCT Pub. No.: WO2010/066203 OTHER PUBLICATIONS PCT Pub. Date: Jun. 17, 2010 Crowley and Zhang, Pharmaceutical Application of Hot Melt Extru (65) Prior Publication Data sion: Part I, Drug Development and Industrial Pharmacy, 2007. 33:909-926.* US 2011/024.4043 A1 Oct. 6, 2011 The Use of Poly (L-Lactide) and RGD Modified Microspheres as Cell Carriers in a Flow Intermittency Bioreactor for Tissue Engi (30) Foreign Application Priority Data neering Cartilage. -

Pushing Estrogen Receptor Around in Breast Cancer
Page 1 of 55 Accepted Preprint first posted on 11 October 2016 as Manuscript ERC-16-0427 1 Pushing estrogen receptor around in breast cancer 2 3 Elgene Lim 1,♯, Gerard Tarulli 2,♯, Neil Portman 1, Theresa E Hickey 2, Wayne D Tilley 4 2,♯,*, Carlo Palmieri 3,♯,* 5 6 1Garvan Institute of Medical Research and St Vincent’s Hospital, University of New 7 South Wales, NSW, Australia. 2Dame Roma Mitchell Cancer Research Laboratories 8 and Adelaide Prostate Cancer Research Centre, University of Adelaide, SA, 9 Australia. 3Institute of Translational Medicine, University of Liverpool, Clatterbridge 10 Cancer Centre, NHS Foundation Trust, and Royal Liverpool University Hospital, 11 Liverpool, UK. 12 13 ♯These authors contributed equally. *To whom correspondence should be addressed: 14 [email protected] or [email protected] 15 16 Short title: Pushing ER around in Breast Cancer 17 18 Keywords: Estrogen Receptor; Endocrine Therapy; Endocrine Resistance; Breast 19 Cancer; Progesterone receptor; Androgen receptor; 20 21 Word Count: 5620 1 Copyright © 2016 by the Society for Endocrinology. Page 2 of 55 22 Abstract 23 The Estrogen receptor-α (herein called ER) is a nuclear sex steroid receptor (SSR) 24 that is expressed in approximately 75% of breast cancers. Therapies that modulate 25 ER action have substantially improved the survival of patients with ER-positive breast 26 cancer, but resistance to treatment still remains a major clinical problem. Treating 27 resistant breast cancer requires co-targeting of ER and alternate signalling pathways 28 that contribute to resistance to improve the efficacy and benefit of currently available 29 treatments. -

Effect of Tamoxifen Or Anastrozole on Steroid Sulfatase Activity and Serum Androgen Concentrations in Postmenopausal Women with Breast Cancer
ANTICANCER RESEARCH 31: 1367-1372 (2011) Effect of Tamoxifen or Anastrozole on Steroid Sulfatase Activity and Serum Androgen Concentrations in Postmenopausal Women with Breast Cancer S.J. STANWAY1, C. PALMIERI2, F.Z. STANCZYK3, E.J. FOLKERD4, M. DOWSETT4, R. WARD2, R.C. COOMBES2, M.J. REED1† and A. PUROHIT1 1Oncology Drug Discovery Group, Section of Investigative Medicine, Imperial College London, Hammersmith Hospital, London W12 0NN, U.K.; 2Cancer Research UK Laboratories, Department of Oncology, Hammersmith Hospital, London W12 0NN, U.K.; 3Reproductive Endocrine Research Laboratory, University of Southern California, Keck School of Medicine, Women’s and Children’s Hospital, Los Angeles, CA, U.S.A.; 4Department of Biochemistry, Royal Marsden Hospital, London, SW3 6JJ, U.K. Abstract. Background: In postmenopausal women sulfate and dehydroepiandrosterone levels. Results: Neither estrogens can be formed by the aromatase pathway, which anastrozole nor tamoxifen had any significant effect on STS gives rise to estrone, and the steroid sulfatase (STS) route activity as measured in PBLs. Anastrozole did not affect which can result in the formation of estrogens and serum androstenediol concentrations. Conclusion: androstenediol, a steroid with potent estrogenic properties. Anastrozole and tamoxifen did not inhibit STS activity and Aromatase inhibitors, such as anastrozole, are now in serum androstenediol concentrations were not reduced by clinical use whereas STS inhibitors, such as STX64, are still aromatase inhibition. As androstenediol has estrogenic undergoing clinical evaluation. STX64 was recently shown properties, it is possible that the combination of an to block STS activity and reduce serum androstenediol aromatase inhibitor and STS inhibitor may give a therapeutic concentrations in postmenopausal women with breast cancer. -

REVIEW Steroid Sulfatase Inhibitors for Estrogen
99 REVIEW Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers Atul Purohit and Paul A Foster1 Oncology Drug Discovery Group, Section of Investigative Medicine, Imperial College London, Hammersmith Hospital, London W12 0NN, UK 1School of Clinical and Experimental Medicine, Centre for Endocrinology, Diabetes and Metabolism, University of Birmingham, Birmingham B15 2TT, UK (Correspondence should be addressed to P A Foster; Email: [email protected]) Abstract Estrogens and androgens are instrumental in the maturation of in vivo and where we currently stand in regards to clinical trials many hormone-dependent cancers. Consequently,the enzymes for these drugs. STS inhibitors are likely to play an important involved in their synthesis are cancer therapy targets. One such future role in the treatment of hormone-dependent cancers. enzyme, steroid sulfatase (STS), hydrolyses estrone sulfate, Novel in vivo models have been developed that allow pre-clinical and dehydroepiandrosterone sulfate to estrone and dehydroe- testing of inhibitors and the identification of lead clinical piandrosterone respectively. These are the precursors to the candidates. Phase I/II clinical trials in postmenopausal women formation of biologically active estradiol and androstenediol. with breast cancer have been completed and other trials in This review focuses on three aspects of STS inhibitors: patients with hormone-dependent prostate and endometrial 1) chemical development, 2) biological activity, and 3) clinical cancer are currently active. Potent STS inhibitors should trials. The aim is to discuss the importance of estrogens and become therapeutically valuable in hormone-dependent androgens in many cancers, the developmental history of STS cancers and other non-oncological conditions. -

Preferred Drug List 4-Tier
Preferred Drug List 4-Tier 21NVHPN13628 Four-Tier Base Drug Benefit Guide Introduction As a member of a health plan that includes outpatient prescription drug coverage, you have access to a wide range of effective and affordable medications. The health plan utilizes a Preferred Drug List (PDL) (also known as a drug formulary) as a tool to guide providers to prescribe clinically sound yet cost-effective drugs. This list was established to give you access to the prescription drugs you need at a reasonable cost. Your out- of-pocket prescription cost is lower when you use preferred medications. Please refer to your Prescription Drug Benefit Rider or Evidence of Coverage for specific pharmacy benefit information. The PDL is a list of FDA-approved generic and brand name medications recommended for use by your health plan. The list is developed and maintained by a Pharmacy and Therapeutics (P&T) Committee comprised of actively practicing primary care and specialty physicians, pharmacists and other healthcare professionals. Patient needs, scientific data, drug effectiveness, availability of drug alternatives currently on the PDL and cost are all considerations in selecting "preferred" medications. Due to the number of drugs on the market and the continuous introduction of new drugs, the PDL is a dynamic and routinely updated document screened regularly to ensure that it remains a clinically sound tool for our providers. Reading the Drug Benefit Guide Benefits for Covered Drugs obtained at a Designated Plan Pharmacy are payable according to the applicable benefit tiers described below, subject to your obtaining any required Prior Authorization or meeting any applicable Step Therapy requirement. -

Antimicrotubule Effects of Estramustine, an Antiprostatic Tumor Drug
[CANCER RESEARCH 45, 3891-3897, August 1985] Antimicrotubule Effects of Estramustine, an Antiprostatic Tumor Drug Mark E. Stearns1 and Kenneth D. Tew2 Departments of Anatomy [M. E. S.] and of Medicine and Pharmacology [K. D, T.], Vincent T. Lombardi Cancer Research Center, Georgetown University Hospital, Washington, DC 20007 ABSTRACT cytomatrix effects of EM and learn how cytoplasmic related effects of EM might ultimately produce the reported antimitotic Estramustine [170-estradiol 3 N bis(2-chloroethyl)carbamate; events in dividing cells (6). EM] is a stable conjugate of estradiol and nor-nitrogen mustard In this paper, we have investigated possible cytotoxic effects that is used for the treatment of human prostatic carcinoma. We of EM at the cytological level. For these studies, the fish erythro- have studied the cytotoxic effects of EM on the cytoskeletal phore or red pigment cell has been used as a model system for organization of squirrelfish pigment cells (erythrophores) and investigation of the cytotoxic consequences of EM. There are a human prostatic tumor cells (DU 145) in culture. Light and whole- number of attractive reasons for utilizing erythrophores for the mount electron microscopy studies reveal that, at /¿Mlevels(60 work described here. Erythrophores are symmetrical cells with to 120 ¿IM),EMhas a dose-dependent disruptive effect on cell thousands of radially ordered microtubules which control the shape, cytoskeletal organization, and intracellular transport. directed motion of numerous red pigment granules (14, 19). At Upon removal of the drug, the cytological effects of EM are the light microscopic level, the pigment is observed to pulsate or rapidly reversible in fish cells but not DU 145s. -

Order in Council 1243/1995
PROVINCE OF BRITISH COLUMBIA ORDER OF THE LIEUTENANT GOVERNOR IN COUNCIL Order in Council No. 12 4 3 , Approved and Ordered OCT 121995 Lieutenant Governor Executive Council Chambers, Victoria On the recommendation of the undersigned, the Lieutenant Governor, by and with the advice and consent of the Executive Council, orders that Order in Council 1039 made August 17, 1995, is rescinded. 2. The Drug Schedules made by regulation of the Council of the College of Pharmacists of British Columbia, as set out in the attached resolution dated September 6, 1995, are hereby approved. (----, c" g/J1"----c- 4- Minister of Heal fandand Minister Responsible for Seniors Presidin Member of the Executive Council (This pan is for adnwustratlye purposes only and is not part of the Order) Authority under which Order Is made: Act and section:- Pharmacists, Pharmacy Operations and Drug Scheduling Act, section 59(2)(1), 62 Other (specify): - Uppodukoic1enact N6145; Resolution of the Council of the College of Pharmacists of British Columbia ("the Council"), made by teleconference at Vancouver, British Columbia, the 6th day of September 1995. RESOLVED THAT: In accordance with the authority established in Section 62 of the Pharmacists, Pharmacy Operations and Drug Scheduling Act of British Columbia, S.B.C. Chapter 62, the Council makes the Drug Schedules by regulation as set out in the attached schedule, subject to the approval of the Lieutenant Governor in Council. Certified a true copy Linda J. Lytle, Phr.) Registrar DRUG SCHEDULES to the Pharmacists, Pharmacy Operations and Drug Scheduling Act of British Columbia The Drug Schedules have been printed in an alphabetical format to simplify the process of locating each individual drug entry and determining its status in British Columbia. -

Emcyt® Estramustine Phosphate Sodium Capsules DESCRIPTION
Emcyt® estramustine phosphate sodium capsules DESCRIPTION Estramustine phosphate sodium, an antineoplastic agent, is an off-white powder readily soluble in water. EMCYT Capsules are white and opaque, each containing estramustine phosphate sodium as the disodium salt monohydrate equivalent to 140 mg estramustine phosphate, for oral administration. Each capsule also contains magnesium stearate, silicon dioxide, sodium lauryl sulfate, and talc. Gelatin capsule shells contain the following pigment: titanium dioxide. Chemically, estramustine phosphate sodium is estra-1,3,5(10)-triene-3,17-diol(17ß)-,3 [bis(2-chloroethyl)carbamate] 17-(dihydrogen phosphate), disodium salt, monohydrate. It is also referred to as estradiol 3-[bis(2-chloroethyl)carbamate] 17-(dihydrogen phosphate), disodium salt, monohydrate. Estramustine phosphate sodium has an empiric formula of C23H30Cl2NNa2O6P•H2O, a calculated molecular weight of 582.4, and the following structural formula: CLINICAL PHARMACOLOGY Estramustine phosphate (Figure 1) is a molecule combining estradiol and nornitrogen mustard by a carbamate link. The molecule is phosphorylated to make it water soluble. 1 Estramustine phosphate taken orally is readily dephosphorylated during absorption, and the major metabolites in plasma are estramustine (Figure 2), the estrone analog (Figure 3), estradiol, and estrone. Prolonged treatment with estramustine phosphate produces elevated total plasma concentrations of estradiol that fall within ranges similar to the elevated estradiol levels found in prostatic cancer patients given conventional estradiol therapy. Estrogenic effects, as demonstrated by changes in circulating levels of steroids and pituitary hormones, are similar in patients treated with either estramustine phosphate or conventional estradiol. 2 The metabolic urinary patterns of the estradiol moiety of estramustine phosphate and estradiol itself are very similar, although the metabolites derived from estramustine phosphate are excreted at a slower rate. -

2021 Formulary List of Covered Prescription Drugs
2021 Formulary List of covered prescription drugs This drug list applies to all Individual HMO products and the following Small Group HMO products: Sharp Platinum 90 Performance HMO, Sharp Platinum 90 Performance HMO AI-AN, Sharp Platinum 90 Premier HMO, Sharp Platinum 90 Premier HMO AI-AN, Sharp Gold 80 Performance HMO, Sharp Gold 80 Performance HMO AI-AN, Sharp Gold 80 Premier HMO, Sharp Gold 80 Premier HMO AI-AN, Sharp Silver 70 Performance HMO, Sharp Silver 70 Performance HMO AI-AN, Sharp Silver 70 Premier HMO, Sharp Silver 70 Premier HMO AI-AN, Sharp Silver 73 Performance HMO, Sharp Silver 73 Premier HMO, Sharp Silver 87 Performance HMO, Sharp Silver 87 Premier HMO, Sharp Silver 94 Performance HMO, Sharp Silver 94 Premier HMO, Sharp Bronze 60 Performance HMO, Sharp Bronze 60 Performance HMO AI-AN, Sharp Bronze 60 Premier HDHP HMO, Sharp Bronze 60 Premier HDHP HMO AI-AN, Sharp Minimum Coverage Performance HMO, Sharp $0 Cost Share Performance HMO AI-AN, Sharp $0 Cost Share Premier HMO AI-AN, Sharp Silver 70 Off Exchange Performance HMO, Sharp Silver 70 Off Exchange Premier HMO, Sharp Performance Platinum 90 HMO 0/15 + Child Dental, Sharp Premier Platinum 90 HMO 0/20 + Child Dental, Sharp Performance Gold 80 HMO 350 /25 + Child Dental, Sharp Premier Gold 80 HMO 250/35 + Child Dental, Sharp Performance Silver 70 HMO 2250/50 + Child Dental, Sharp Premier Silver 70 HMO 2250/55 + Child Dental, Sharp Premier Silver 70 HDHP HMO 2500/20% + Child Dental, Sharp Performance Bronze 60 HMO 6300/65 + Child Dental, Sharp Premier Bronze 60 HDHP HMO