Syntabulin Regulates the Trafficking of PICK1-Containing Vesicles in Neurons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GOPC-ROS1 Fusion Due to Microdeletion at 6Q22 Is an Oncogenic Driver in a Subset of Pediatric Gliomas And
J Neuropathol Exp Neurol Vol. 78, No. 12, December 2019, pp. 1089–1099 doi: 10.1093/jnen/nlz093 ORIGINAL ARTICLE GOPC-ROS1 Fusion Due to Microdeletion at 6q22 Is an Oncogenic Driver in a Subset of Pediatric Gliomas and Glioneuronal Tumors Downloaded from https://academic.oup.com/jnen/article/78/12/1089/5593615 by New York University user on 25 August 2020 Timothy E. Richardson, DO, PhD, Karen Tang, MD, Varshini Vasudevaraja, MS, Jonathan Serrano, BS, Christopher M. William, MD, PhD, Kanish Mirchia, MD, Christopher R. Pierson, MD, PhD, Jeffrey R. Leonard, MD, Mohamed S. AbdelBaki, MD, Kathleen M. Schieffer, PhD, Catherine E. Cottrell, PhD, Zulma Tovar-Spinoza, MD, Melanie A. Comito, MD, Daniel R. Boue, MD, PhD, George Jour, MD, and Matija Snuderl, MD features; the third tumor aligned best with glioblastoma and Abstract showed no evidence of neuronal differentiation. Copy number ROS1 is a transmembrane receptor tyrosine kinase proto- profiling revealed chromosome 6q22 microdeletions correspond- oncogene that has been shown to have rearrangements with sev- ingtotheGOPC-ROS1 fusion in all 3 cases and methylation pro- eral genes in glioblastoma and other neoplasms, including intra- filing showed that the tumors did not cluster together as a single chromosomal fusion with GOPC due to microdeletions at 6q22.1. entity or within known methylation classes by t-Distributed Sto- ROS1 fusion events are important findings in these tumors, as chastic Neighbor Embedding. they are potentially targetable alterations with newer tyrosine ki- nase inhibitors; however, whether these tumors represent a dis- Key Words: 6q22, Astrocytoma, Brain tumor, GOPC, Pediatric gli- tinct entity remains unknown. -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
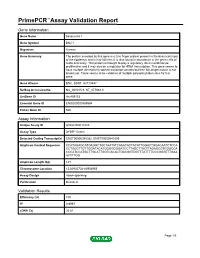
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Comparative Transcriptome Profiling of Selected Osmotic Regulatory Proteins in the Gill During Seawater Acclimation of Chum Salm
www.nature.com/scientificreports OPEN Comparative transcriptome profling of selected osmotic regulatory proteins in the gill during seawater acclimation of chum salmon (Oncorhynchus keta) fry Sang Yoon Lee 1, Hwa Jin Lee2 & Yi Kyung Kim1,2* Salmonid fshes, chum salmon (Oncorhynchus keta) have the developed adaptive strategy to withstand wide salinity changes from the early life stage. This study investigated gene expression patterns of cell membrane proteins in the gill of chum salmon fry on the transcriptome level by tracking the salinity acclimation of the fsh in changing environments ranging from freshwater (0 ppt) to brackish water (17.5 ppt) to seawater (35 ppt). Using GO analysis of DEGs, the known osmoregulatory genes and their functional groups such as ion transport, transmembrane transporter activity and metal ion binding were identifed. The expression patterns of membrane protein genes, including pump-mediated protein (NKA, CFTR), carrier-mediated protein (NKCC, NHE3) and channel-mediated protein (AQP) were similar to those of other salmonid fshes in the smolt or adult stages. Based on the protein-protein interaction analysis between transmembrane proteins and other related genes, we identifed osmotic-related genes expressed with salinity changes and analyzed their expression patterns. The fndings of this study may facilitate the disentangling of the genetic basis of chum salmon and better able an understanding of the osmophysiology of the species. Salinity is one of the critical factors limiting the distribution patterns of all aquatic organisms1–4. Salmonid fshes display diverse life-history traits; anadromous individuals that mature in the river from hatching through to juveniles acquire the capacity to tolerate salinity associated with parr–smolt transformation and undergo ocean migrations before returning to rivers for spawning, whereas landlocked types spend their entire life within fresh- water5,6. -

Rna-Sequencing Applications: Gene Expression Quantification and Methylator Phenotype Identification
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 8-2013 RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION Guoshuai Cai Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Cai, Guoshuai, "RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 386. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/386 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION -

The GALNT9, BNC1 and CCDC8 Genes Are Frequently Epigenetically Dysregulated in Breast Tumours That Metastasise to the Brain Rajendra P
Pangeni et al. Clinical Epigenetics (2015) 7:57 DOI 10.1186/s13148-015-0089-x RESEARCH Open Access The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain Rajendra P. Pangeni1, Prasanna Channathodiyil1, David S. Huen2, Lawrence W. Eagles1, Balraj K. Johal2, Dawar Pasha2, Natasa Hadjistephanou2, Oliver Nevell2, Claire L. Davies2, Ayobami I. Adewumi2, Hamida Khanom2, Ikroop S. Samra2, Vanessa C. Buzatto2, Preethi Chandrasekaran2, Thoraia Shinawi3, Timothy P. Dawson4, Katherine M. Ashton4, Charles Davis4, Andrew R. Brodbelt5, Michael D. Jenkinson5, Ivan Bièche6, Farida Latif3, John L. Darling1, Tracy J. Warr1 and Mark R. Morris1,2,3* Abstract Background: Tumour metastasis to the brain is a common and deadly development in certain cancers; 18–30 % of breast tumours metastasise to the brain. The contribution that gene silencing through epigenetic mechanisms plays in these metastatic tumours is not well understood. Results: We have carried out a bioinformatic screen of genome-wide breast tumour methylation data available at The Cancer Genome Atlas (TCGA) and a broad literature review to identify candidate genes that may contribute to breast to brain metastasis (BBM). This analysis identified 82 candidates. We investigated the methylation status of these genes using Combined Bisulfite and Restriction Analysis (CoBRA) and identified 21 genes frequently methylated in BBM. We have identified three genes, GALNT9, CCDC8 and BNC1, that were frequently methylated (55, 73 and 71 %, respectively) and silenced in BBM and infrequently methylated in primary breast tumours. CCDC8 was commonly methylated in brain metastases and their associated primary tumours whereas GALNT9 and BNC1 were methylated and silenced only in brain metastases, but not in the associated primary breast tumours from individual patients. -

Identification of Candidate Tumour Suppressor Genes Frequently
Oncogene (2010) 29, 2104–2117 & 2010 Macmillan Publishers Limited All rights reserved 0950-9232/10 $32.00 www.nature.com/onc ORIGINAL ARTICLE Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma MR Morris1,2, C Ricketts1,2, D Gentle1,2, M Abdulrahman2, N Clarke3, M Brown3, T Kishida4, M Yao4, F Latif1,2 and ER Maher1,2,5 1Cancer Research UK Renal Molecular Oncology Group, University of Birmingham, Birmingham, UK; 2Department of Medical and Molecular Genetics, School of Clinical and Experimental Medicine, College of Medical and Dental Sciences, University of Birmingham, Birmingham, UK; 3Genito-Urinary Cancer Research Group, Paterson Institute for Cancer Research, University of Manchester, Manchester, UK; 4Department of Urology, Yokohama City University School of Medicine, Yokohama, Japan and 5West Midlands Region Genetics Service, Birmingham Women’s Hospital, Edgbaston, Birmingham, UK Promoter region hyermethylation and transcriptional cancer are diagnosed in the world each year (Ferlay silencing is a frequent cause of tumour suppressor gene et al., 2007). The most common form of kidney cancer in (TSG) inactivation in many types of human cancers. adults is renal cell carcinoma (RCC). A majority of the Functional epigenetic studies, in which gene expression is RCC (B75%) are classified as clear cell (conventional) induced by treatment with demethylating agents, may and the next most frequent subtype is papillary RCC identify novel genes with tumour-specific methylation. We (B15% of all cases) (Mancini et al., 2008). The used high-density gene expression microarrays in a prognosis of advanced RCC is poor, although newer functional epigenetic study of 11 renal cell carcinoma treatments, based on knowledge of the molecular (RCC) cell lines.