Genomic Anatomy of the Tyrp1 (Brown) Deletion Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
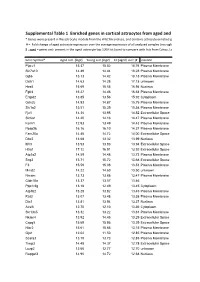
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

2007-05 TLE1 Synovial Sarcoma
TLE1 Immunostains in the Diagnosis of Synovial Sarcoma May 2007 by Rodney T. Miller, M.D., Director of Immunohistochemistry The diagnosis of synovial sarcoma can be a chal- lenging task, particularly on small biopsy specimens, as the morphologic features of this tumor can be mimicked by a variety of other neoplasms. This month we call attention to a paper published in the February 2007 edition of the American Journal of Surgical Pathology describing the utility of immu- nostains for TLE1 in the diagnosis of this tumor. TLE immunostains are now available at ProPath. Synovial sarcoma occurs in three morphologic varie- ties: monophasic, biphasic, and poorly differentiated. It has been known for some time that synovial sar- coma is associated with a specific chromosomal H&E (left) and TLE1 immunostain (right) on a monophasic translocation, t(X;18), that results in the fusion of the synovial sarcoma. Note the numerous strongly positive nuclei SYT gene on chromosome 18 to either the SSX1 or on the TLE1 immunostain, a typical feature of this tumor. SSX2 gene on the X chromosome, resulting in the production of a SYT-SSX fusion protein. Identifica- In their study, the authors performed TLE1 immu- tion of this translocation in the appropriate setting is nostains on multiple tissue microarrays using two dif- regarded by many to be diagnostic of synovial sar- ferent antibodies (monoclonal and polyclonal), both coma. However, the methodologies used for this performing in a similar fashion. A total of 693 adult purpose (cytogenetics, fluorescent in situ hybridiza- soft tissue tumors were examined, including 94 cases tion, and reverse-transcriptase polymerase chain re- of synovial sarcoma that had documentation of the t action) are not readily available in many diagnostic (X;18) translocation. -
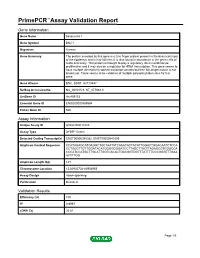
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Nuclear Factor I/B Is an Oncogene in Small Cell Lung Cancer
Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Dooley, A. L. et al. “Nuclear factor I/B is an oncogene in small cell lung cancer.” Genes & Development 25 (2011): 1470-1475. As Published http://dx.doi.org/10.1101/gad.2046711 Publisher Cold Spring Harbor Laboratory Press in association with The Genetics Society Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/66512 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Dooley 1 Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer Alison L. Dooley1, Monte M. Winslow1, Derek Y. Chiang2,3,4, Shantanu Banerji2,3, Nicolas Stransky2, Talya L. Dayton1, Eric L. Snyder1, Stephanie Senna1, Charles A. Whittaker1, Roderick T. Bronson5, Denise Crowley1, Jordi Barretina2,3, Levi Garraway2,3, Matthew Meyerson2,3, Tyler Jacks1,6 1David H. Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2The Broad Institute, Cancer Program, Cambridge, Massachusetts, USA 3Dana-Farber Cancer Institute, Department of Medical Oncology and Center for Cancer Genome Discovery, Boston, Massachusetts, USA 4Current address: Lineberger Comprehensive Cancer Center, 450 West Drive, CB #7295, Chapel Hill, North Carolina, USA 5Department of Pathology, Tufts University School of Medicine and Veterinary Medicine, North Grafton, Massachusetts, USA 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA Key Words: Small Cell Lung Cancer, Mouse model, Nuclear Factor I/B Dooley 2 Abstract Small cell lung cancer (SCLC) is an aggressive cancer often diagnosed after it has metastasized. -

Multi-Class Protein Classification Using Adaptive Codes
Journal of Machine Learning Research 8 (2007) 1557-1581 Submitted 8/06; Revised 4/07; Published 7/07 Multi-class Protein Classification Using Adaptive Codes Iain Melvin∗ [email protected] NEC Laboratories of America Princeton, NJ 08540, USA Eugene Ie∗ [email protected] Department of Computer Science and Engineering University of California San Diego, CA 92093-0404, USA Jason Weston [email protected] NEC Laboratories of America Princeton, NJ 08540, USA William Stafford Noble [email protected] Department of Genome Sciences Department of Computer Science and Engineering University of Washington Seattle, WA 98195, USA Christina Leslie [email protected] Center for Computational Learning Systems Columbia University New York, NY 10115, USA Editor: Nello Cristianini Abstract Predicting a protein’s structural class from its amino acid sequence is a fundamental problem in computational biology. Recent machine learning work in this domain has focused on develop- ing new input space representations for protein sequences, that is, string kernels, some of which give state-of-the-art performance for the binary prediction task of discriminating between one class and all the others. However, the underlying protein classification problem is in fact a huge multi- class problem, with over 1000 protein folds and even more structural subcategories organized into a hierarchy. To handle this challenging many-class problem while taking advantage of progress on the binary problem, we introduce an adaptive code approach in the output space of one-vs- the-rest prediction scores. Specifically, we use a ranking perceptron algorithm to learn a weight- ing of binary classifiers that improves multi-class prediction with respect to a fixed set of out- put codes. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B
Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B by Geoffrey Graham Lundell-Smith A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Biochemistry University of Toronto © Copyright by Geoffrey Lundell-Smith, 2017 Characterizing Novel Interactions of Transcriptional Repression Proteins BCL6 and BCL6B Geoffrey Graham Lundell-Smith Masters of Science Department of Biochemistry University of Toronto 2016 Abstract B-cell Lymphoma 6 (BCL6) and its close homolog BCL6B encode proteins that are members of the BTB-Zinc Finger family of transcription factors. BCL6 plays an important role in regulating the differentiation and proliferation of B-cells during the adaptive immune response, and is also involved in T cell development and inflammation. BCL6 acts by repressing genes involved in DNA damage response during the affinity maturation of immunoglobulins, and the mis- expression of BCL6 can lead to diffuse large B-cell lymphoma. Although BCL6B shares high sequence similarity with BCL6, the functions of BCL6B are not well-characterized. I used BioID, an in vivo proximity-dependent labeling method, to identify novel BCL6 and BCL6B protein interactors and validated a number of these interactions with co-purification experiments. I also examined the evolutionary relationship between BCL6 and BCL6B and identified conserved residues in an important interaction interface that mediates corepressor binding and gene repression. ii Acknowledgments Thank you to my supervisor, Gil Privé for his mentorship, guidance, and advice, and for giving me the opportunity to work in his lab. Thanks to my committee members, Dr. John Rubinstein and Dr. Jeff Lee for their ideas, thoughts, and feedback during my Masters. -

Establishing Incentives and Changing Cultures to Support Data Access
EXPERT ADVISORY GROUP ON DATA ACCESS ESTABLISHING INCENTIVES AND CHANGING CULTURES TO SUPPORT DATA ACCESS May 2014 ACKNOWLEDGEMENT This is a report of the Expert Advisory Group on Data Access (EAGDA). EAGDA was established by the MRC, ESRC, Cancer Research UK and the Wellcome Trust in 2012 to provide strategic advice on emerging scientific, ethical and legal issues in relation to data access for cohort and longitudinal studies. The report is based on work undertaken by the EAGDA secretariat at the Group’s request. The contributions of David Carr, Natalie Banner, Grace Gottlieb, Joanna Scott and Katherine Littler at the Wellcome Trust are gratefully acknowledged. EAGDA would also like to thank the representatives of the MRC, ESRC and Cancer Research UK for their support and input throughout the project. Most importantly, EAGDA owes a considerable debt of gratitude to the many individuals from the research community who contributed to this study through feeding in their expert views via surveys, interviews and focus groups. The Expert Advisory Group on Data Access Martin Bobrow (Chair) Bartha Maria Knoppers James Banks Mark McCarthy Paul Burton Andrew Morris George Davey Smith Onora O'Neill Rosalind Eeles Nigel Shadbolt Paul Flicek Chris Skinner Mark Guyer Melanie Wright Tim Hubbard 1 EXECUTIVE SUMMARY This project was developed as a key component of the workplan of the Expert Advisory Group on Data Access (EAGDA). EAGDA wished to understand the factors that help and hinder individual researchers in making their data (both published and unpublished) available to other researchers, and to examine the potential need for new types of incentives to enable data access and sharing. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol .