Genomic Anatomy of the Tyrp1 (Brown) Deletion Complex
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gene Section Short Communication
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL INIST-CNRS Gene Section Short Communication TYRP1 (tyrosinase-related protein 1) Kunal Ray, Mainak Sengupta, Sampurna Ghosh Academy of Scientific and Innovative Research (AcSIR), Campus at CSIR - Central Road Research Institute, Mathura Road, New Delhi - 110 025, [email protected] (KR); University of Calcutta, Department of Genetics, 35, Ballygunge Circular Road, Kolkata - 700 019, [email protected]); [email protected] (MS, SG) India. Published in Atlas Database: April 2016 Online updated version : http://AtlasGeneticsOncology.org/Genes/TYRP1ID46370ch9p23.html Printable original version : http://documents.irevues.inist.fr/bitstream/handle/2042/68125/04-2016-TYRP1ID46370ch9p23.pdf DOI: 10.4267/2042/68125 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2016 Atlas of Genetics and Cytogenetics in Oncology and Haematology Abstract Location: 9p23 TYRP1 gene, having a chromosomal location of 9p23, encodes a melanosomal enzyme belonging to DNA/RNA the tyrosinase family. TYRP1 catalyses oxidation of 5,6-dihydroxyindole-2-carboxylic acid (DHICA) Description into indole-5,6-quinone-2-carboxylic acid. TYRP1 In Chromosome 9, the 24,852 bases long gene starts is also thought to play a role in stabilizing tyrosinase from12,685,439 bp from pter and ends at 12,710,290 and modulates its catalytic activity, in maintenance bp from pter; Orientation: Plus strand. The gene of melanosome structure, affecting melanocyte contains 8 exons and spans ~24.8 kb of the genome. proliferation and melanocyte cell death. Defects in this gene cause oculocutaneous albinism type III; Transcription OCA III (also known as rufous oculocutaneous The gene encodes a 2876 bp mRNA. -

Dog Coat Colour Genetics: a Review Date Published Online: 31/08/2020; 1,2 1 1 3 Rashid Saif *, Ali Iftekhar , Fatima Asif , Mohammad Suliman Alghanem
www.als-journal.com/ ISSN 2310-5380/ August 2020 Review Article Advancements in Life Sciences – International Quarterly Journal of Biological Sciences ARTICLE INFO Open Access Date Received: 02/05/2020; Date Revised: 20/08/2020; Dog Coat Colour Genetics: A Review Date Published Online: 31/08/2020; 1,2 1 1 3 Rashid Saif *, Ali Iftekhar , Fatima Asif , Mohammad Suliman Alghanem Authors’ Affiliation: 1. Institute of Abstract Biotechnology, Gulab Devi Educational anis lupus familiaris is one of the most beloved pet species with hundreds of world-wide recognized Complex, Lahore - Pakistan breeds, which can be differentiated from each other by specific morphological, behavioral and adoptive 2. Decode Genomics, traits. Morphological characteristics of dog breeds get more attention which can be defined mostly by 323-D, Town II, coat color and its texture, and considered to be incredibly lucrative traits in this valued species. Although Punjab University C Employees Housing the genetic foundation of coat color has been well stated in the literature, but still very little is known about the Scheme, Lahore - growth pattern, hair length and curly coat trait genes. Skin pigmentation is determined by eumelanin and Pakistan 3. Department of pheomelanin switching phenomenon which is under the control of Melanocortin 1 Receptor and Agouti Signaling Biology, Tabuk Protein genes. Genetic variations in the genes involved in pigmentation pathway provide basic understanding of University - Kingdom melanocortin physiology and evolutionary adaptation of this trait. So in this review, we highlighted, gathered and of Saudi Arabia comprehend the genetic mutations, associated and likely to be associated variants in the genes involved in the coat color and texture trait along with their phenotypes. -

Chromosomal Aberrations in Head and Neck Squamous Cell Carcinomas in Norwegian and Sudanese Populations by Array Comparative Genomic Hybridization
825-843 12/9/08 15:31 Page 825 ONCOLOGY REPORTS 20: 825-843, 2008 825 Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization ERIC ROMAN1,2, LEONARDO A. MEZA-ZEPEDA3, STINE H. KRESSE3, OLA MYKLEBOST3,4, ENDRE N. VASSTRAND2 and SALAH O. IBRAHIM1,2 1Department of Biomedicine, Faculty of Medicine and Dentistry, University of Bergen, Jonas Lies vei 91; 2Department of Oral Sciences - Periodontology, Faculty of Medicine and Dentistry, University of Bergen, Årstadveien 17, 5009 Bergen; 3Department of Tumor Biology, Institute for Cancer Research, Rikshospitalet-Radiumhospitalet Medical Center, Montebello, 0310 Oslo; 4Department of Molecular Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway Received January 30, 2008; Accepted April 29, 2008 DOI: 10.3892/or_00000080 Abstract. We used microarray-based comparative genomic logical parameters showed little correlation, suggesting an hybridization to explore genome-wide profiles of chromosomal occurrence of gains/losses regardless of ethnic differences and aberrations in 26 samples of head and neck cancers compared clinicopathological status between the patients from the two to their pair-wise normal controls. The samples were obtained countries. Our findings indicate the existence of common from Sudanese (n=11) and Norwegian (n=15) patients. The gene-specific amplifications/deletions in these tumors, findings were correlated with clinicopathological variables. regardless of the source of the samples or attributed We identified the amplification of 41 common chromosomal carcinogenic risk factors. regions (harboring 149 candidate genes) and the deletion of 22 (28 candidate genes). Predominant chromosomal alterations Introduction that were observed included high-level amplification at 1q21 (harboring the S100A gene family) and 11q22 (including Head and neck squamous cell carcinoma (HNSCC), including several MMP family members). -
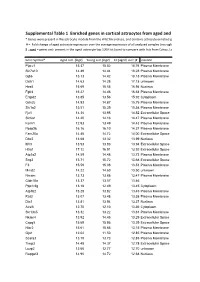
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

Two Locus Inheritance of Non-Syndromic Midline Craniosynostosis Via Rare SMAD6 and 4 Common BMP2 Alleles 5 6 Andrew T
1 2 3 Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and 4 common BMP2 alleles 5 6 Andrew T. Timberlake1-3, Jungmin Choi1,2, Samir Zaidi1,2, Qiongshi Lu4, Carol Nelson- 7 Williams1,2, Eric D. Brooks3, Kaya Bilguvar1,5, Irina Tikhonova5, Shrikant Mane1,5, Jenny F. 8 Yang3, Rajendra Sawh-Martinez3, Sarah Persing3, Elizabeth G. Zellner3, Erin Loring1,2,5, Carolyn 9 Chuang3, Amy Galm6, Peter W. Hashim3, Derek M. Steinbacher3, Michael L. DiLuna7, Charles 10 C. Duncan7, Kevin A. Pelphrey8, Hongyu Zhao4, John A. Persing3, Richard P. Lifton1,2,5,9 11 12 1Department of Genetics, Yale University School of Medicine, New Haven, CT, USA 13 2Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, CT, USA 14 3Section of Plastic and Reconstructive Surgery, Department of Surgery, Yale University School of Medicine, New Haven, CT, USA 15 4Department of Biostatistics, Yale University School of Medicine, New Haven, CT, USA 16 5Yale Center for Genome Analysis, New Haven, CT, USA 17 6Craniosynostosis and Positional Plagiocephaly Support, New York, NY, USA 18 7Department of Neurosurgery, Yale University School of Medicine, New Haven, CT, USA 19 8Child Study Center, Yale University School of Medicine, New Haven, CT, USA 20 9The Rockefeller University, New York, NY, USA 21 22 ABSTRACT 23 Premature fusion of the cranial sutures (craniosynostosis), affecting 1 in 2,000 24 newborns, is treated surgically in infancy to prevent adverse neurologic outcomes. To 25 identify mutations contributing to common non-syndromic midline (sagittal and metopic) 26 craniosynostosis, we performed exome sequencing of 132 parent-offspring trios and 59 27 additional probands. -

Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Genetic basis of simple and complex traits with relevance to avian evolution Małgorzata Anna Gazda Doctoral Program in Biodiversity, Genetics and Evolution D Faculdade de Ciências da Universidade do Porto 2019 Supervisor Miguel Jorge Pinto Carneiro, Auxiliary Researcher, CIBIO/InBIO, Laboratório Associado, Universidade do Porto Co-supervisor Ricardo Lopes, CIBIO/InBIO Leif Andersson, Uppsala University FCUP Genetic basis of avian traits Nota Previa Na elaboração desta tese, e nos termos do número 2 do Artigo 4º do Regulamento Geral dos Terceiros Ciclos de Estudos da Universidade do Porto e do Artigo 31º do D.L.74/2006, de 24 de Março, com a nova redação introduzida pelo D.L. 230/2009, de 14 de Setembro, foi efetuado o aproveitamento total de um conjunto coerente de trabalhos de investigação já publicados ou submetidos para publicação em revistas internacionais indexadas e com arbitragem científica, os quais integram alguns dos capítulos da presente tese. Tendo em conta que os referidos trabalhos foram realizados com a colaboração de outros autores, o candidato esclarece que, em todos eles, participou ativamente na sua conceção, na obtenção, análise e discussão de resultados, bem como na elaboração da sua forma publicada. Este trabalho foi apoiado pela Fundação para a Ciência e Tecnologia (FCT) através da atribuição de uma bolsa de doutoramento (PD/BD/114042/2015) no âmbito do programa doutoral em Biodiversidade, Genética e Evolução (BIODIV). 2 FCUP Genetic basis of avian traits Acknowledgements Firstly, I would like to thank to my all supervisors Miguel Carneiro, Ricardo Lopes and Leif Andersson, for the demanding task of supervising myself last four years. -

Nuclear Factor I/B Is an Oncogene in Small Cell Lung Cancer
Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Dooley, A. L. et al. “Nuclear factor I/B is an oncogene in small cell lung cancer.” Genes & Development 25 (2011): 1470-1475. As Published http://dx.doi.org/10.1101/gad.2046711 Publisher Cold Spring Harbor Laboratory Press in association with The Genetics Society Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/66512 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Dooley 1 Nuclear Factor I/B is an Oncogene in Small Cell Lung Cancer Alison L. Dooley1, Monte M. Winslow1, Derek Y. Chiang2,3,4, Shantanu Banerji2,3, Nicolas Stransky2, Talya L. Dayton1, Eric L. Snyder1, Stephanie Senna1, Charles A. Whittaker1, Roderick T. Bronson5, Denise Crowley1, Jordi Barretina2,3, Levi Garraway2,3, Matthew Meyerson2,3, Tyler Jacks1,6 1David H. Koch Institute for Integrative Cancer Research and Department of Biology, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA 2The Broad Institute, Cancer Program, Cambridge, Massachusetts, USA 3Dana-Farber Cancer Institute, Department of Medical Oncology and Center for Cancer Genome Discovery, Boston, Massachusetts, USA 4Current address: Lineberger Comprehensive Cancer Center, 450 West Drive, CB #7295, Chapel Hill, North Carolina, USA 5Department of Pathology, Tufts University School of Medicine and Veterinary Medicine, North Grafton, Massachusetts, USA 6Howard Hughes Medical Institute, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA Key Words: Small Cell Lung Cancer, Mouse model, Nuclear Factor I/B Dooley 2 Abstract Small cell lung cancer (SCLC) is an aggressive cancer often diagnosed after it has metastasized. -

Review of the Current State of Genetic Testing - a Living Resource
Review of the Current State of Genetic Testing - A Living Resource Prepared by Liza Gershony, DVM, PhD and Anita Oberbauer, PhD of the University of California, Davis Editorial input by Leigh Anne Clark, PhD of Clemson University July, 2020 Contents Introduction .................................................................................................................................................. 1 I. The Basics ......................................................................................................................................... 2 II. Modes of Inheritance ....................................................................................................................... 7 a. Mendelian Inheritance and Punnett Squares ................................................................................. 7 b. Non-Mendelian Inheritance ........................................................................................................... 10 III. Genetic Selection and Populations ................................................................................................ 13 IV. Dog Breeds as Populations ............................................................................................................. 15 V. Canine Genetic Tests ...................................................................................................................... 16 a. Direct and Indirect Tests ................................................................................................................ 17 b. Single -

Original Article a Database and Functional Annotation of NF-Κb Target Genes
Int J Clin Exp Med 2016;9(5):7986-7995 www.ijcem.com /ISSN:1940-5901/IJCEM0019172 Original Article A database and functional annotation of NF-κB target genes Yang Yang, Jian Wu, Jinke Wang The State Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, People’s Republic of China Received November 4, 2015; Accepted February 10, 2016; Epub May 15, 2016; Published May 30, 2016 Abstract: Backgrounds: The previous studies show that the transcription factor NF-κB always be induced by many inducers, and can regulate the expressions of many genes. The aim of the present study is to explore the database and functional annotation of NF-κB target genes. Methods: In this study, we manually collected the most complete listing of all NF-κB target genes identified to date, including the NF-κB microRNA target genes and built the database of NF-κB target genes with the detailed information of each target gene and annotated it by DAVID tools. Results: The NF-κB target genes database was established (http://tfdb.seu.edu.cn/nfkb/). The collected data confirmed that NF-κB maintains multitudinous biological functions and possesses the considerable complexity and diversity in regulation the expression of corresponding target genes set. The data showed that the NF-κB was a central regula- tor of the stress response, immune response and cellular metabolic processes. NF-κB involved in bone disease, immunological disease and cardiovascular disease, various cancers and nervous disease. NF-κB can modulate the expression activity of other transcriptional factors. Inhibition of IKK and IκBα phosphorylation, the decrease of nuclear translocation of p65 and the reduction of intracellular glutathione level determined the up-regulation or down-regulation of expression of NF-κB target genes. -

Genetic Analysis of Rare Human Variants of Regulators of G Protein Signaling Proteins and Their Role in Human Physiology and Diseases
Supplemental Material can be found at: /content/suppl/2018/06/05/70.3.446.DC1.html 1521-0081/70/3/446–474$35.00 https://doi.org/10.1124/pr.117.015354 PHARMACOLOGICAL REVIEWS Pharmacol Rev 70:446–474, July 2018 Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics ASSOCIATE EDITOR: ALAN V. SMRCKA Genetic Analysis of Rare Human Variants of Regulators of G Protein Signaling Proteins and Their Role in Human Physiology and Diseases Katherine E. Squires, Carolina Montañez-Miranda, Rushika R. Pandya, Matthew P. Torres, and John R. Hepler Department of Pharmacology, Emory University School of Medicine, Atlanta, Georgia (K.E.S., C.M.-M., J.R.H.); and School of Biological Sciences, Georgia Institute of Technology, Atlanta, Georgia (R.R.P., M.P.T.) Abstract. ....................................................................................446 I. Introduction. ..............................................................................447 A. G Protein Signaling......................................................................447 B. A (Very) Brief History of Regulators of G Protein Signaling...............................447 C. RGS Protein Rare Human Variants in Complex Diseases . ...............................448 II. RGS Proteins in Physiology and Human Disease . ..........................................449 A. The R4 Family . ........................................................................449 Downloaded from 1. R4 Family Overview..................................................................449 -

RASEF Expression Correlates with Hormone Receptor Status in Breast Cancer
ONCOLOGY LETTERS 16: 7223-7230, 2018 RASEF expression correlates with hormone receptor status in breast cancer MASAHIRO SHIBATA1, MITSURO KANDA2, DAI SHIMIZU2, HARUYOSHI TANAKA2, SHINICHI UMEDA2, TAKASHI MIWA2, MASAMICHI HAYASHI2, TAKAHIRO INAISHI1, NORIYUKI MIYAJIMA1, YAYOI ADACHI1, YUKO TAKANO1, KENICHI NAKANISHI1, DAI TAKEUCHI1, SUMIYO NODA1, YASUHIRO KODERA2 and TOYONE KIKUMORI1 Departments of 1Breast and Endocrine Surgery (Surgery II) and 2Gastroenterological Surgery (Surgery II), Nagoya University Graduate School of Medicine, Nagoya, Aichi 466-8550, Japan Received February 26, 2018; Accepted September 25, 2018 DOI: 10.3892/ol.2018.9542 Abstract. Breast cancer (BC) is the most frequently estrogen receptor status (P<0.001), negative progesterone diagnosed malignant tumor in women worldwide, and the receptor status (P<0.001), and triple-negative status (P<0.001). development of new molecules associated with BC is essential Additionally, although the differences were not statistically for the management of this disease. RAS and EF-hand significant, patients with low RASEF expression levels domain-containing (RASEF) encodes the GTPase enzyme exhibited poorer disease-free survival (P=0.123) and overall that belongs to the Rab family. Although the effects of this survival (P=0.086) than other patients. The results of the gene have been reported in several malignant tumor types, the present study indicate that RASEF mRNA expression levels role of RASEF in BC has not been completely elucidated. The are associated with hormone receptor status in BC. aim of the present study was to investigate the importance of RASEF expression in BC. RASEF mRNA expression levels Introduction were evaluated in BC and non-cancerous mammary cell lines.