Genetic Basis of Simple and Complex Traits with Relevance to Avian Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Novel Approach to Identify Driver Genes Involved in Androgen-Independent Prostate Cancer
Schinke et al. Molecular Cancer 2014, 13:120 http://www.molecular-cancer.com/content/13/1/120 RESEARCH Open Access A novel approach to identify driver genes involved in androgen-independent prostate cancer Ellyn N Schinke1, Victor Bii1, Arun Nalla1, Dustin T Rae1, Laura Tedrick1, Gary G Meadows1 and Grant D Trobridge1,2* Abstract Background: Insertional mutagenesis screens have been used with great success to identify oncogenes and tumor suppressor genes. Typically, these screens use gammaretroviruses (γRV) or transposons as insertional mutagens. However, insertional mutations from replication-competent γRVs or transposons that occur later during oncogenesis can produce passenger mutations that do not drive cancer progression. Here, we utilized a replication-incompetent lentiviral vector (LV) to perform an insertional mutagenesis screen to identify genes in the progression to androgen-independent prostate cancer (AIPC). Methods: Prostate cancer cells were mutagenized with a LV to enrich for clones with a selective advantage in an androgen-deficient environment provided by a dysregulated gene(s) near the vector integration site. We performed our screen using an in vitro AIPC model and also an in vivo xenotransplant model for AIPC. Our approach identified proviral integration sites utilizing a shuttle vector that allows for rapid rescue of plasmids in E. coli that contain LV long terminal repeat (LTR)-chromosome junctions. This shuttle vector approach does not require PCR amplification and has several advantages over PCR-based techniques. Results: Proviral integrations were enriched near prostate cancer susceptibility loci in cells grown in androgen- deficient medium (p < 0.001), and five candidate genes that influence AIPC were identified; ATPAF1, GCOM1, MEX3D, PTRF, and TRPM4. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -
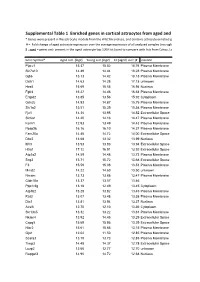
Supplemental Table 1 Enriched Genes in Cortical Astrocytes from Aged
Supplemental Table 1 Enriched genes in cortical astrocytes from aged and young-adult mice * Genes were present in the astrocyte module from the WGCNA analysis, and contains astrocyte enriched genes compared to microglia and oligodendrocytes # = Fold change of aged astrocyte expression over the average expression of all analyzed samples (microglia, astrocytes: young, old, with and without myelin contamination) $ ; aged = genes only present in the aged astrocyte top 1000 list (used to compare with lists from Cahoy, Lovatt, Doyle; see Fig. 4B), all = genes present in all astrocyte top 1000 lists Gene Symbol* Aged astr. (log2) Young astr.(log2) FC (aged/ aver.)# Location Ptprz1 15.37 15.02 18.76 Plasma Membrane Slc7a10 14.49 14.44 18.28 Plasma Membrane Gjb6 15.13 14.42 18.18 Plasma Membrane Dclk1 14.63 14.28 17.18 unknown Hes5 15.69 15.55 16.94 Nucleus Fgfr3 15.27 14.46 16.54 Plasma Membrane Entpd2 13.85 13.56 15.92 Cytoplasm Grin2c 14.93 14.87 15.75 Plasma Membrane Slc1a2 15.51 15.39 15.58 Plasma Membrane Fjx1 14.36 13.98 14.52 Extracellular Space Slc6a1 14.20 14.16 14.47 Plasma Membrane Kcnk1 12.93 13.49 14.43 Plasma Membrane Ppap2b 16.16 16.10 14.37 Plasma Membrane Fam20a 14.48 14.72 14.00 Extracellular Space Dbx2 13.68 13.32 13.99 Nucleus Itih3 13.93 13.93 13.94 Extracellular Space Htra1 17.12 16.91 13.92 Extracellular Space Atp1a2 14.59 14.48 13.73 Plasma Membrane Scg3 15.71 15.72 13.68 Extracellular Space F3 15.59 15.08 13.51 Plasma Membrane Mmd2 14.22 14.60 13.50 unknown Nrcam 13.73 13.88 13.47 Plasma Membrane Cldn10a 13.37 13.57 13.46 -

New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology
Cancer Research and Treatment 2003;35(4):304-313 New Approaches to Functional Process Discovery in HPV 16-Associated Cervical Cancer Cells by Gene Ontology Yong-Wan Kim, Ph.D.1, Min-Je Suh, M.S.1, Jin-Sik Bae, M.S.1, Su Mi Bae, M.S.1, Joo Hee Yoon, M.D.2, Soo Young Hur, M.D.2, Jae Hoon Kim, M.D.2, Duck Young Ro, M.D.2, Joon Mo Lee, M.D.2, Sung Eun Namkoong, M.D.2, Chong Kook Kim, Ph.D.3 and Woong Shick Ahn, M.D.2 1Catholic Research Institutes of Medical Science, 2Department of Obstetrics and Gynecology, College of Medicine, The Catholic University of Korea, Seoul; 3College of Pharmacy, Seoul National University, Seoul, Korea Purpose: This study utilized both mRNA differential significant genes of unknown function affected by the display and the Gene Ontology (GO) analysis to char- HPV-16-derived pathway. The GO analysis suggested that acterize the multiple interactions of a number of genes the cervical cancer cells underwent repression of the with gene expression profiles involved in the HPV-16- cancer-specific cell adhesive properties. Also, genes induced cervical carcinogenesis. belonging to DNA metabolism, such as DNA repair and Materials and Methods: mRNA differential displays, replication, were strongly down-regulated, whereas sig- with HPV-16 positive cervical cancer cell line (SiHa), and nificant increases were shown in the protein degradation normal human keratinocyte cell line (HaCaT) as a con- and synthesis. trol, were used. Each human gene has several biological Conclusion: The GO analysis can overcome the com- functions in the Gene Ontology; therefore, several func- plexity of the gene expression profile of the HPV-16- tions of each gene were chosen to establish a powerful associated pathway, identify several cancer-specific cel- cervical carcinogenesis pathway. -

An Abridged Compendium of Words. a Discussion of Them and Opinions About Them
DERMATOLOGY PRACTICAL & CONCEPTUAL www.derm101.com Dermatopathology: An abridged compendium of words. A discussion of them and opinions about them. Part 6 (I-L) Bruce J. Hookerman1 1 Dermatology Specialists, Bridgeton, Missouri, USA Citation: Hookerman BJ. Dermatopathology: An abridged compendium of words. A discussion of them and opinions about them. Part 6 (I-L). Dermatol Pract Concept. 2014;4(4):1. http://dx.doi.org/10.5826/dpc.0404a01 Copyright: ©2014 Hookerman. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Corresponding author: Bruce J. Hookerman, M.D., 12105 Bridgeton Square Drive, St. Louis, MO 63044, USA. Email: [email protected] – I – term “id reaction” only for a spongiotic dermatitis manifested by tiny vesicles on the hands of patients with florid dermato- ICHTHYOSIS: a generic term for skin conditions character- phytosis at another site, usually the feet, or for an analogue ized by what are said to be fishlike scales, i.e., scales that are of that phenomenon such as widespread vesicles that appear broad and polygonal with free edges, as are seen in ichthyosis subsequent to injudicious treatment, i.e., with Gentian violet vulgaris (and its look-alike, acquired ichthyosis), X-linked (known sardonically in times past as “Gentian violent”) of ichthyosis, and lamellar ichthyosis. Conditions reputed to be an exuberant spongiotic dermatitis, usually on the feet, such ichthyosis, such as ichthyosis hystrix and ichthyosis linearis as an allergic contact dermatitis. A time-honored explana- circumflexa, do not qualify because they are not associated tion for an “id” reaction is hematogenous dissemination of with broad polygonal scales. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Phylogeography of Finches and Sparrows
In: Animal Genetics ISBN: 978-1-60741-844-3 Editor: Leopold J. Rechi © 2009 Nova Science Publishers, Inc. Chapter 1 PHYLOGEOGRAPHY OF FINCHES AND SPARROWS Antonio Arnaiz-Villena*, Pablo Gomez-Prieto and Valentin Ruiz-del-Valle Department of Immunology, University Complutense, The Madrid Regional Blood Center, Madrid, Spain. ABSTRACT Fringillidae finches form a subfamily of songbirds (Passeriformes), which are presently distributed around the world. This subfamily includes canaries, goldfinches, greenfinches, rosefinches, and grosbeaks, among others. Molecular phylogenies obtained with mitochondrial DNA sequences show that these groups of finches are put together, but with some polytomies that have apparently evolved or radiated in parallel. The time of appearance on Earth of all studied groups is suggested to start after Middle Miocene Epoch, around 10 million years ago. Greenfinches (genus Carduelis) may have originated at Eurasian desert margins coming from Rhodopechys obsoleta (dessert finch) or an extinct pale plumage ancestor; it later acquired green plumage suitable for the greenfinch ecological niche, i.e.: woods. Multicolored Eurasian goldfinch (Carduelis carduelis) has a genetic extant ancestor, the green-feathered Carduelis citrinella (citril finch); this was thought to be a canary on phonotypical bases, but it is now included within goldfinches by our molecular genetics phylograms. Speciation events between citril finch and Eurasian goldfinch are related with the Mediterranean Messinian salinity crisis (5 million years ago). Linurgus olivaceus (oriole finch) is presently thriving in Equatorial Africa and was included in a separate genus (Linurgus) by itself on phenotypical bases. Our phylograms demonstrate that it is and old canary. Proposed genus Acanthis does not exist. Twite and linnet form a separate radiation from redpolls. -

Elevated Levels of the Steroidogenic Factor 1 Are Associated with Over
European Journal of Endocrinology (2012) 166 941–949 ISSN 0804-4643 CASE REPORT Elevated levels of the steroidogenic factor 1 are associated with over-expression of CYP19 in an oestrogen-producing testicular Leydig cell tumour Anne Hege Straume1,2, Kristian Løva˚s3,4, Hrvoje Miletic5,6, Karsten Gravdal5, Per Eystein Lønning1,2 and Stian Knappskog1,2 1Section of Oncology, Institute of Medicine, University of Bergen, Bergen, Norway, 2Department of Oncology, Haukeland University Hospital, Bergen, Norway, 3Section of Endocrinology, Institute of Medicine, University of Bergen, Bergen, Norway, 4Department of Medicine and 5Section of Pathology, Haukeland University Hospital, Bergen, Norway and 6Department of Biomedicine, University of Bergen, Bergen, Norway (Correspondence should be addressed to S Knappskog who is now at Mohn Cancer Research Laboratory (1M), Haukeland University Hospital, 5021 Bergen, Norway; Email: [email protected]) Abstract Background and objectives: Testicular Leydig cell tumours (LCTs) are rare, steroid-secreting tumours. Elevated levels of aromatase (CYP19 or CYP19A1) mRNA have been previously described in LCTs; however, little is known about the mechanism(s) causing CYP19 over-expression. We report an LCT in a 29-year-old male with elevated plasma oestradiol caused by enhanced CYP19 transcription. Design and methods: First, we measured the intra-tumour expression of CYP19 and determined the use of CYP19 promoters by qPCR. Secondly, we explored CYP19 and promoter II (PII) for gene amplifications and activating mutations in PII by sequencing. Thirdly, we analysed intra-tumour expression of steroidogenic factor 1 (SF-1 (NR5A1)), liver receptor homologue-1 (LRH-1 (NR5A2)) and cyclooxygenase-2 (COX2 (PTGS2)). Finally, we analysed SF-1 for promoter mutations and gene amplifications. -

A General Binding Mechanism for All Human Sulfatases by the Formylglycine-Generating Enzyme
A general binding mechanism for all human sulfatases by the formylglycine-generating enzyme Dirk Roeser*, Andrea Preusser-Kunze†, Bernhard Schmidt†, Kathrin Gasow*, Julia G. Wittmann*, Thomas Dierks‡, Kurt von Figura†, and Markus Georg Rudolph*§ *Department of Molecular Structural Biology, University of Go¨ttingen, Justus-von-Liebig-Weg 11, D-37077 Go¨ttingen, Germany; †Department of Biochemistry II, Heinrich-Du¨ker-Weg 12, University of Go¨ttingen, D-37073 Go¨ttingen, Germany; and ‡Department of Biochemistry I, Universita¨tsstrasse 25, University of Bielefeld, D-33615 Bielefeld, Germany Edited by Carolyn R. Bertozzi, University of California, Berkeley, CA, and approved November 8, 2005 (received for review September 1, 2005) The formylglycine (FGly)-generating enzyme (FGE) uses molecular tases, suggesting a general binding mechanism of substrate sulfa- oxygen to oxidize a conserved cysteine residue in all eukaryotic tases by FGE. sulfatases to the catalytically active FGly. Sulfatases degrade and The details of how O2-dependent cysteine oxidation is mediated remodel sulfate esters, and inactivity of FGE results in multiple by FGE are unknown. As a first step toward the elucidation of the sulfatase deficiency, a fatal disease. The previously determined FGE molecular mechanism of FGly formation, we have previously crystal structure revealed two crucial cysteine residues in the active determined crystal structures of FGE in various oxidation states site, one of which was thought to be implicated in substrate (8). FGE adopts a novel fold with surprisingly little regular sec- 2ϩ binding. The other cysteine residue partakes in a novel oxygenase ondary structure and contains two structural Ca ions and two mechanism that does not rely on any cofactors. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

Lysosomal Hydrolases: Conversion of Acidic to Basic Forms by Neuraminidase
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Volume 13, number 1 FEBS LETTERS February 1971 LYSOSOMAL HYDROLASES: CONVERSION OF ACIDIC TO BASIC FORMS BY NEURAMINIDASE Alfred GOLDSTONE, Philip KONECNY and Harold KOENIG Neurology Service, VA Research Hospital; and Departments of Neurology and Biochemistry, Northwestern University Medical School, Chicago, Illinois, USA Received 21 December 1970 1. Introduction intervals to avoid overheating). The lysosomal lysate was ultracentrifuged at 100,000 g for 30 min to give A number of hydrolytic enzymes of lysosomal a soluble enzyme fraction. This fraction also contains origin occur in multiple forms which differ in such an acidic lipoprotein [4]. The insoluble residue was physicochemical properties as solubility, heat and pH suspended in 0.05 M acetate buffer pH 5.0 with 0.2% stability, pH optima and electrophoretic mobility [I]. Triton X-100 and resonicated to release the bound ly- Recent studies in this laboratory indicate that essen- sosomal enzymes together with additional acidic lipo- tially all the enzyme proteins of rat kidney and liver protein ([4] and A. Goldstone and H. Koenig, unpu- lysosomes are glycoproteins, and that at least some of blished observations). All operations were carried out these glycoproteins contain sialic acid [2-41. The at 4’. acidic form of lysosomal N-acetylhexosaminidase from Aliquots of the soluble fraction containing about human spleen [ 51 and kidney [6] is converted into 2 mg of protein were incubated with neuraminidase the basic form by treatment with neuraminidase. We from Cl. perfringens (Type Vl enzyme supplied by now confirm this finding for rat kidney and show Sigma), 0.1 mg/ml, or V.