The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice
Loyola University Chicago Loyola eCommons Biology: Faculty Publications and Other Works Faculty Publications 2013 Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice Mihaela Palicev Gunter P. Wagner James P. Noonan Benedikt Hallgrimsson James M. Cheverud Loyola University Chicago, [email protected] Follow this and additional works at: https://ecommons.luc.edu/biology_facpubs Part of the Biology Commons Recommended Citation Palicev, M, GP Wagner, JP Noonan, B Hallgrimsson, and JM Cheverud. "Genomic Correlates of Relationship QTL Involved in Fore- Versus Hind Limb Divergence in Mice." Genome Biology and Evolution 5(10), 2013. This Article is brought to you for free and open access by the Faculty Publications at Loyola eCommons. It has been accepted for inclusion in Biology: Faculty Publications and Other Works by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. © Palicev et al., 2013. GBE Genomic Correlates of Relationship QTL Involved in Fore- versus Hind Limb Divergence in Mice Mihaela Pavlicev1,2,*, Gu¨ nter P. Wagner3, James P. Noonan4, Benedikt Hallgrı´msson5,and James M. Cheverud6 1Konrad Lorenz Institute for Evolution and Cognition Research, Altenberg, Austria 2Department of Pediatrics, Cincinnati Children‘s Hospital Medical Center, Cincinnati, Ohio 3Yale Systems Biology Institute and Department of Ecology and Evolutionary Biology, Yale University 4Department of Genetics, Yale University School of Medicine 5Department of Cell Biology and Anatomy, The McCaig Institute for Bone and Joint Health and the Alberta Children’s Hospital Research Institute for Child and Maternal Health, University of Calgary, Calgary, Canada 6Department of Anatomy and Neurobiology, Washington University *Corresponding author: E-mail: [email protected]. -

AN INVESTIGATION of the ROLE of PAK6 in TUMORIGENESIS By
AN INVESTIGATION OF THE ROLE OF PAK6 IN TUMORIGENESIS by JoAnn Roberts A Thesis Submitted to the Faculty of The Charles E. Schmidt College of Medicine In Partial Fulfillment of the Requirements for the Degree of Master of Science Florida Atlantic University Boca Raton, Florida August 2012 ACKNOWLEDGMENTS This material is based upon work supported by the National Science Foundation under Grant No. DGE: 0638662. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. I would like to thank and acknowledge my thesis advisor, Dr. Michael Lu, for his support and guidance throughout the writing of this thesis and design of experiments in this manuscript. I would also like to thank my colleagues for assistance in various trouble-shooting circumstances. Last, but certainly not least, I would like to thank my family and friends for their support in the pursuit of my graduate studies. iii ABSTRACT Author: JoAnn Roberts Title: An Investigation of the Role of PAK6 in Tumorigenesis Institution: Florida Atlantic University Thesis Advisor: Dr. Michael Lu Degree: Master of Science Year: 2012 The function and role of PAK6, a serine/threonine kinase, in cancer progression has not yet been clearly identified. Several studies reveal that PAK6 may participate in key changes contributing to cancer progression such as cell survival, cell motility, and invasiveness. Based on the membrane localization of PAK6 in prostate and breast cancer cells, we speculated that PAK6 plays a role in cancer progression cells by localizing on the membrane and modifying proteins linked to motility and proliferation. -
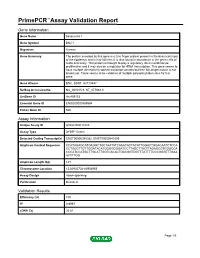
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

PIK3C2G (NM 004570) Human Mutant ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC402758 PIK3C2G (NM_004570) Human Mutant ORF Clone Product data: Product Type: Mutant ORF Clones Product Name: PIK3C2G (NM_004570) Human Mutant ORF Clone Mutation Description: P146L Affected Codon#: 146 Affected NT#: 437 Nucleotide Mutation: PIK3C2G Mutant (P146L), Myc-DDK-tagged ORF clone of Homo sapiens phosphoinositide-3- kinase, class 2, gamma polypeptide (PIK3C2G) as transfection-ready DNA Effect: Dibees, ype 2, ssoiion wih Symbol: PIK3C2G Synonyms: PI3K-C2-gamma; PI3K-C2GAMMA Vector: pCMV6-Entry (PS100001) Tag: Myc-DDK ACCN: NM_004570 ORF Size: 4335 bp ORF Nucleotide >RC402758 representing NM_004570 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCATATTCTTGGCAAACGGATCCAAATCCTAATGAATCACACGAAAAGCAGTATGAACACCAAGAAT TTCTCTTTGTAAATCAACCCCATTCTTCTAGCCAAGTCAGTCTGGGTTTTGATCAGATAGTAGATGAGAT CAGTGGCAAAATTCCACACTACGAGAGTGAAATTGATGAAAACACCTTTTTTGTGCCCACTGCACCAAAA TGGGACTCAACAGGGCATTCATTAAATGAAGCACACCAAATATCCTTGAATGAATTCACTTCTAAAAGCC GTGAACTCTCCTGGCATCAAGTTAGCAAAGCACCAGCAATTGGTTTTAGTCCTTCTGTGTTACCAAAACC TCAAAATACGAATAAAGAATGCTCCTGGGGAAGCCCCATAGGAAAACATCATGGTGCTGATGATTCCAGA TTCAGTATTTTAGCTCTATCATTCACAAGTTTGGATAAAATTAATCTAGAGAAAGAATTAGAAAATGAAA ATCATAACTACCATATAGGATTTGAAAGTAGCATTCCTCCAACAAATTCATCCTTCTCAAGTGACTTCAT GCCGAAAGAAGAGAATAAAAGGAGTGGACATGTGAACATTGTGGAACCATCTTTGATGCTTTTGAAAGGC -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

NDRG2 Combined with EGFR Improved Its Prognostic Value for Lung Adenocarcinoma
NDRG2 combined with EGFR improved its prognostic value for lung adenocarcinoma Bo Yang ( [email protected] ) Tianjin First Central Hospital https://orcid.org/0000-0003-0052-048X Xiao-Ping Li Tianjin First Central Hospital Hong-Gang Zhou Nankai University College of Pharmacy Tao Jiang Fourth Military Medical University Ting Xiao Nankai University College of Pharmacy Yu-Li Wei Nankai University College of Pharmacy Liang Zhang Tianjin First Central Hospital Wen-Chen Wang PLA Army General Hospital Wei-Dong Zhang Tianjin First Central Hospital Research article Keywords: N-Myc downstream-regulated gene2; Epidermal growth factor receptor; Adenocarcinoma; Prognosis Posted Date: September 10th, 2019 DOI: https://doi.org/10.21203/rs.2.14181/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/10 Abstract Background N-Myc downstream-regulated gene2 (NDRG2) plays an important role in lung adenocarcinoma (LUAD). Epidermal growth factor receptor (EGFR) mutation has signicantly improved prognosis in patients with adenocarcinoma. We aimed to elucidate the clinical value of NDRG2/EGFR as a prediction of prognosis in patients with lung adenocarcinoma. Materials and Methods Immunohistochemistry and western blot analysis were conducted to detect the expression of NDRG2 protein. Association between NDRG2/EGFR expression and clinicopathological parameters of the patients were examined. Serum Carcinoembryonic antigen (CEA) level was examined prior to treatment in patients with LUAD. Patients’ survival rate was assessed by Kaplan–Meier. Candidates for independent prognostic biomarkers were analyzed using a COX proportional hazard model. Results NDRG2 levels were signicantly decreased in patients with lung adenocarcinoma. NDRG2 levels were positively correlated with CEA and EGFR. -

Analysis of Inherited and Somatic Variants to Decipher Canine Complex Traits
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1454 Analysis of inherited and somatic variants to decipher canine complex traits KATE MEGQUIER ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6206 ISBN 978-91-513-0310-9 UPPSALA urn:nbn:se:uu:diva-347165 2018 Dissertation presented at Uppsala University to be publicly examined in B:22, BMC, Husargatan 3, Uppsala, Monday, 21 May 2018 at 13:15 for the degree of Doctor of Philosophy (Faculty of Medicine). The examination will be conducted in English. Faculty examiner: David Sargan (University of Cambridge). Abstract Megquier, K. 2018. Analysis of inherited and somatic variants to decipher canine complex traits. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1454. 67 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0310-9. This thesis presents several investigations of the dog as a model for complex diseases, focusing on cancers and the effect of genetic risk factors on clinical presentation. In Papers I and II, we performed genome-wide association studies (GWAS) to identify germline risk factors predisposing US golden retrievers to hemangiosarcoma (HSA) and B- cell lymphoma (BLSA). Paper I identified two loci predisposing to both HSA and BLSA, approximately 4 megabases (Mb) apart on chromosome 5. Carrying the risk haplotype at these loci was associated with separate changes in gene expression, both relating to T-cell activation and proliferation. Paper II followed up on the HSA GWAS by performing a meta-analysis with additional cases and controls. This confirmed three previously reported GWAS loci for HSA and revealed three new loci, the most significant on chromosome 18. -

Beyond Traditional Morphological Characterization of Lung
Cancers 2020 S1 of S15 Beyond Traditional Morphological Characterization of Lung Neuroendocrine Neoplasms: In Silico Study of Next-Generation Sequencing Mutations Analysis across the Four World Health Organization Defined Groups Giovanni Centonze, Davide Biganzoli, Natalie Prinzi, Sara Pusceddu, Alessandro Mangogna, Elena Tamborini, Federica Perrone, Adele Busico, Vincenzo Lagano, Laura Cattaneo, Gabriella Sozzi, Luca Roz, Elia Biganzoli and Massimo Milione Table S1. Genes Frequently mutated in Typical Carcinoids (TCs). Mutation Original Entrez Gene Gene Rate % eukaryotic translation initiation factor 1A X-linked [Source: HGNC 4.84 EIF1AX 1964 EIF1AX Symbol; Acc: HGNC: 3250] AT-rich interaction domain 1A [Source: HGNC Symbol;Acc: HGNC: 4.71 ARID1A 8289 ARID1A 11110] LDL receptor related protein 1B [Source: HGNC Symbol; Acc: 4.35 LRP1B 53353 LRP1B HGNC: 6693] 3.53 NF1 4763 NF1 neurofibromin 1 [Source: HGNC Symbol;Acc: HGNC: 7765] DS cell adhesion molecule like 1 [Source: HGNC Symbol; Acc: 2.90 DSCAML1 57453 DSCAML1 HGNC: 14656] 2.90 DST 667 DST dystonin [Source: HGNC Symbol;Acc: HGNC: 1090] FA complementation group D2 [Source: HGNC Symbol; Acc: 2.90 FANCD2 2177 FANCD2 HGNC: 3585] piccolo presynaptic cytomatrix protein [Source: HGNC Symbol; Acc: 2.90 PCLO 27445 PCLO HGNC: 13406] erb-b2 receptor tyrosine kinase 2 [Source: HGNC Symbol; Acc: 2.44 ERBB2 2064 ERBB2 HGNC: 3430] BRCA1 associated protein 1 [Source: HGNC Symbol; Acc: HGNC: 2.35 BAP1 8314 BAP1 950] capicua transcriptional repressor [Source: HGNC Symbol; Acc: 2.35 CIC 23152 CIC HGNC: -

Rna-Sequencing Applications: Gene Expression Quantification and Methylator Phenotype Identification
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 8-2013 RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION Guoshuai Cai Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Cai, Guoshuai, "RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 386. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/386 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION -

Integrative Genomic Analysis Identifies NDRG2 As a Candidate Tumor Suppressor Gene Frequently Inactivated in Clinically Aggressive Meningioma
Research Article Integrative Genomic Analysis Identifies NDRG2 as a Candidate Tumor Suppressor Gene Frequently Inactivated in Clinically Aggressive Meningioma Eriks A. Lusis,1 Mark A. Watson,2 Michael R. Chicoine,3 Meghan Lyman,4 Peter Roerig,5 Guido Reifenberger,5 David H. Gutmann,4 and Arie Perry1 1Division of Neuropathology, Departments of 2Pathology and Immunology, 3Neurosurgery, and 4Neurology, Washington University School of Medicine, St. Louis, Missouri; and 5Department of Neuropathology, Heinrich-Heine-University, Du¨sseldorf, Germany Abstract and TSLC1 are also common (2, 4–6). However, there are few Although meningiomas are common central nervous system known genetic changes associated with the malignant progression tumors, little is known about the genetic events responsible of meningioma. Although losses of chromosomal arms 14q and 1p for malignant progression. In this study, we employed gene are common in anaplastic tumors (WHO grade III) and are expression profiling to identify transcripts whose expression generally associated with poor prognosis (7, 8), the relevant tumor was lost in anaplastic (WHO grade III) versus benign (WHO suppressor genes that map to these loci have not been identified. grade I) meningioma. Approximately 40% of genes down- Loss of heterozygosity studies have had limited success in regulated in anaplastic meningioma were localized to chro- identifying minimal regions of deletion, in part, due to the fact mosomes 1p and 14q. One specific gene located at 14q11.2, that the entire chromosome or chromosomal arm is lost in most NDRG2, was consistently down-regulated in grade III menin- examples. In the current study, we used a strategy combining expression profiling with known cytogenetic alterations, leading gioma, a finding which we validated at both the transcript and protein levels in independent sets of clinically and patholog- to the identification of NDRG2 as a potential meningioma- ically diverse meningiomas. -

TRIM24 As an Oncogene in the Mammary Gland
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 5-2018 TRIM24 as an Oncogene in the Mammary Gland Aundrietta Duncan Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Cancer Biology Commons, Medicine and Health Sciences Commons, and the Other Genetics and Genomics Commons Recommended Citation Duncan, Aundrietta, "TRIM24 as an Oncogene in the Mammary Gland" (2018). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 845. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/845 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. TRIM24 AS AN ONCOGENE IN THE MAMMARY GLAND by Aundrietta DeVan Duncan, M.S. APPROVED: ______________________________ Michelle C. Barton, Ph.D. Advisory Professor ______________________________ Richard -

The GALNT9, BNC1 and CCDC8 Genes Are Frequently Epigenetically Dysregulated in Breast Tumours That Metastasise to the Brain Rajendra P
Pangeni et al. Clinical Epigenetics (2015) 7:57 DOI 10.1186/s13148-015-0089-x RESEARCH Open Access The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain Rajendra P. Pangeni1, Prasanna Channathodiyil1, David S. Huen2, Lawrence W. Eagles1, Balraj K. Johal2, Dawar Pasha2, Natasa Hadjistephanou2, Oliver Nevell2, Claire L. Davies2, Ayobami I. Adewumi2, Hamida Khanom2, Ikroop S. Samra2, Vanessa C. Buzatto2, Preethi Chandrasekaran2, Thoraia Shinawi3, Timothy P. Dawson4, Katherine M. Ashton4, Charles Davis4, Andrew R. Brodbelt5, Michael D. Jenkinson5, Ivan Bièche6, Farida Latif3, John L. Darling1, Tracy J. Warr1 and Mark R. Morris1,2,3* Abstract Background: Tumour metastasis to the brain is a common and deadly development in certain cancers; 18–30 % of breast tumours metastasise to the brain. The contribution that gene silencing through epigenetic mechanisms plays in these metastatic tumours is not well understood. Results: We have carried out a bioinformatic screen of genome-wide breast tumour methylation data available at The Cancer Genome Atlas (TCGA) and a broad literature review to identify candidate genes that may contribute to breast to brain metastasis (BBM). This analysis identified 82 candidates. We investigated the methylation status of these genes using Combined Bisulfite and Restriction Analysis (CoBRA) and identified 21 genes frequently methylated in BBM. We have identified three genes, GALNT9, CCDC8 and BNC1, that were frequently methylated (55, 73 and 71 %, respectively) and silenced in BBM and infrequently methylated in primary breast tumours. CCDC8 was commonly methylated in brain metastases and their associated primary tumours whereas GALNT9 and BNC1 were methylated and silenced only in brain metastases, but not in the associated primary breast tumours from individual patients.