Identification of Candidate Tumour Suppressor Genes Frequently
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
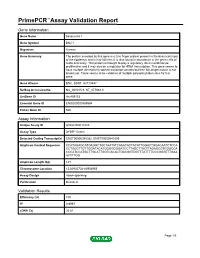
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Characterization of Glyceollins As Novel Aryl Hydrocarbon Receptor Ligands and Their Role in Cell Migration
International Journal of Molecular Sciences Article Characterization of Glyceollins as Novel Aryl Hydrocarbon Receptor Ligands and Their Role in Cell Migration Thu Ha Pham 1, Sylvain Lecomte 1, Remy Le Guevel 2, Aurélie Lardenois 1, Bertrand Evrard 1, Frédéric Chalmel 1, François Ferriere 1, Patrick Balaguer 3 , Theo Efstathiou 4 and Farzad Pakdel 1,* 1 Univ Rennes, Inserm, EHESP, Irset (Institut de recherche en santé, environnement et travail) -UMR_S1085, F-35000 Rennes, France; [email protected] (T.H.P.); [email protected] (S.L.); [email protected] (A.L.); [email protected] (B.E.); [email protected] (F.C.); [email protected] (F.F.) 2 ImPACcell platform (SFR Biosit), Univ Rennes, 35000 Rennes, France; [email protected] 3 Institut de Recherche en Cancérologie de Montpellier (IRCM), INSERM U1194, ICM, Univ. Montpellier, 34090 Montpellier, France; [email protected] 4 Laboratoire Nutrinov, Technopole Atalante Champeaux, 8 Rue Jules Maillard de la Gournerie, 35012 Rennes CEDEX, France; [email protected] * Correspondence: [email protected]; Tel.: +33-(0)22-323-5132 Received: 12 December 2019; Accepted: 14 February 2020; Published: 18 February 2020 Abstract: Recent studies strongly support the use of the aryl hydrocarbon receptor (AhR) as a therapeutic target in breast cancer. Glyceollins, a group of soybean phytoalexins, are known to exert therapeutic effects in chronic human diseases and also in cancer. To investigate the interaction between glyceollin I (GI), glyceollin II (GII) and AhR, a computational docking analysis, luciferase assays, immunofluorescence and transcriptome analyses were performed with different cancer cell lines. -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Gene Expression Profiles Underlying Aggressive Behavior in the Prefrontal Cortex of Cattle Paulina G
Eusebi et al. BMC Genomics (2021) 22:245 https://doi.org/10.1186/s12864-021-07505-5 RESEARCH ARTICLE Open Access Gene expression profiles underlying aggressive behavior in the prefrontal cortex of cattle Paulina G. Eusebi1*, Natalia Sevane1, Thomas O’Rourke2,3, Manuel Pizarro1, Cedric Boeckx2,3,4 and Susana Dunner1 Abstract Background: Aggressive behavior is an ancient and conserved trait, habitual for most animals in order to eat, protect themselves, compete for mating and defend their territories. Genetic factors havebeenshowntoplayanimportantrolein the development of aggression both in animals and humans, displaying moderate to high heritability estimates. Although such types of behaviors have been studied in different animal models, the molecular architecture of aggressiveness remains poorly understood. This study compared gene expression profiles of 16 prefrontal cortex (PFC) samples from aggressive and non-aggressive cattle breeds: Lidia, selected for agonistic responses, and Wagyu, selected for tameness. Results: A total of 918 up-regulated and 278 down-regulated differentially expressed genes (DEG) were identified, representing above-chance overlap with genes previously identified in studies of aggression across species, as well as those implicated in recent human evolution. The functional interpretation of the up-regulated genes in the aggressive cohort revealed enrichment of pathways such as Alzheimer disease-presenilin, integrins and the ERK/MAPK signaling cascade, all implicated in the development of abnormal aggressive behaviors and neurophysiological disorders. Moreover, gonadotropins, are up-regulated as natural mechanisms enhancing aggression. Concomitantly, heterotrimeric G-protein pathways, associated with low reactivity mental states, and the GAD2 gene, a repressor of agonistic reactions associated with PFC activity, are down-regulated, promoting the development of the aggressive responses selected for in Lidia cattle. -

Analysis of the Indacaterol-Regulated Transcriptome in Human Airway
Supplemental material to this article can be found at: http://jpet.aspetjournals.org/content/suppl/2018/04/13/jpet.118.249292.DC1 1521-0103/366/1/220–236$35.00 https://doi.org/10.1124/jpet.118.249292 THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS J Pharmacol Exp Ther 366:220–236, July 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Analysis of the Indacaterol-Regulated Transcriptome in Human Airway Epithelial Cells Implicates Gene Expression Changes in the s Adverse and Therapeutic Effects of b2-Adrenoceptor Agonists Dong Yan, Omar Hamed, Taruna Joshi,1 Mahmoud M. Mostafa, Kyla C. Jamieson, Radhika Joshi, Robert Newton, and Mark A. Giembycz Departments of Physiology and Pharmacology (D.Y., O.H., T.J., K.C.J., R.J., M.A.G.) and Cell Biology and Anatomy (M.M.M., R.N.), Snyder Institute for Chronic Diseases, Cumming School of Medicine, University of Calgary, Calgary, Alberta, Canada Received March 22, 2018; accepted April 11, 2018 Downloaded from ABSTRACT The contribution of gene expression changes to the adverse and activity, and positive regulation of neutrophil chemotaxis. The therapeutic effects of b2-adrenoceptor agonists in asthma was general enriched GO term extracellular space was also associ- investigated using human airway epithelial cells as a therapeu- ated with indacaterol-induced genes, and many of those, in- tically relevant target. Operational model-fitting established that cluding CRISPLD2, DMBT1, GAS1, and SOCS3, have putative jpet.aspetjournals.org the long-acting b2-adrenoceptor agonists (LABA) indacaterol, anti-inflammatory, antibacterial, and/or antiviral activity. Numer- salmeterol, formoterol, and picumeterol were full agonists on ous indacaterol-regulated genes were also induced or repressed BEAS-2B cells transfected with a cAMP-response element in BEAS-2B cells and human primary bronchial epithelial cells by reporter but differed in efficacy (indacaterol $ formoterol . -

Role and Regulation of the P53-Homolog P73 in the Transformation of Normal Human Fibroblasts
Role and regulation of the p53-homolog p73 in the transformation of normal human fibroblasts Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Lars Hofmann aus Aschaffenburg Würzburg 2007 Eingereicht am Mitglieder der Promotionskommission: Vorsitzender: Prof. Dr. Dr. Martin J. Müller Gutachter: Prof. Dr. Michael P. Schön Gutachter : Prof. Dr. Georg Krohne Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig angefertigt und keine anderen als die angegebenen Hilfsmittel und Quellen verwendet habe. Diese Arbeit wurde weder in gleicher noch in ähnlicher Form in einem anderen Prüfungsverfahren vorgelegt. Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren akademischen Grade erworben und zu erwerben gesucht. Würzburg, Lars Hofmann Content SUMMARY ................................................................................................................ IV ZUSAMMENFASSUNG ............................................................................................. V 1. INTRODUCTION ................................................................................................. 1 1.1. Molecular basics of cancer .......................................................................................... 1 1.2. Early research on tumorigenesis ................................................................................. 3 1.3. Developing -

Global Reactivation of Epigenetically Silenced Genes in Prostate Cancer
Published OnlineFirst August 10, 2010; DOI: 10.1158/1940-6207.CAPR-10-0039 Research Article Cancer Prevention See perspective p. 1053 Research Global Reactivation of Epigenetically Silenced Genes in Prostate Cancer Ilsiya Ibragimova1, Inmaculada Ibáñez de Cáceres1, Amanda M. Hoffman1, Anna Potapova1, Essel Dulaimi1, Tahseen Al-Saleem2, Gary R. Hudes3, Michael F. Ochs4, and Paul Cairns1,2 Abstract Transcriptional silencing associated with aberrant promoter hypermethylation is a common mechanism of inactivation of tumor suppressor genes in cancer cells. To globally profile the genes silenced by hypermethylation in prostate cancer, we screened a whole genome expression microarray for genes reactivated in the LNCaP, DU-145, PC-3, and MDA2b prostate tumor cell lines after treatment with the demethylating drug 5-aza-2-deoxycytidine and the histone deacetylation–inhibiting drug trichostatin A. A total of 2,997 genes showed at least 2-fold upregulation of expression after drug treatment in at least one prostate tumor cell line. For validation, we examined the first 45 genes, ranked by upregulation of expression, which had a typical CpG island and were known to be expressed in the normal cell counterpart. Two important findings were, first, that several genes known to be frequently hypermethylated in prostate cancer were apparent, and, second, that validation studies revealed eight novel genes hypermethylated in the prostate tumor cell lines, four of which were unmethylated in normal prostate cells and hypermethylated in primary prostate tumors (SLC15A3, 66%; KRT7, 54%; TACSTD2, 17%; GADD45b, 3%). Thus, we established the utility of our screen for genes hypermethylated in prostate cancer cells. One of the novel genes was TACSTD2/TROP2, a marker of human prostate basal cells with stem cell characteristics. -

View – 2003;22(12):3164 74
Qin et al. Clinical Epigenetics (2015) 7:97 DOI 10.1186/s13148-015-0131-z RESEARCH Open Access Epigenetic synergy between decitabine and platinum derivatives Taichun Qin1†, Jiali Si1†, Noël J-M Raynal1,4, Xiaodan Wang3, Vazganush Gharibyan1, Saira Ahmed1, Xin Hu1, Chunlei Jin1, Yue Lu1,2, Jingmin Shu1, Marcos RH Estecio1,2, Jaroslav Jelinek1,4 and Jean-Pierre J. Issa1,4* Abstract Background: Aberrant epigenetic silencing of tumor suppressor genes has been recognized as a driving force in cancer. Epigenetic drugs such as the DNA methylation inhibitor decitabine reactivate genes and are effective in myeloid leukemia, but resistance often develops and efficacy in solid tumors is limited. To improve their clinical efficacy, we searched among approved anti-cancer drugs for an epigenetic synergistic combination with decitabine. Results: We used the YB5 cell line, a clonal derivative of the SW48 colon cancer cell line that contains a single copy of a hypermethylated cytomegalovirus (CMV) promoter driving green fluorescent protein (GFP) to screen for drug-induced gene reactivation and synergy with decitabine. None of the 16 anti-cancer drugs tested had effects on their own. However, in combination with decitabine, platinum compounds showed striking synergy in activating GFP. This was dose dependent, observed both in concurrent and sequential combinations, and also seen with other alkylating agents. Clinically achievable concentrations of carboplatin at (25 μM) and decitabine reactivated GFP in 28 % of the YB5 cells as compared to 15 % with decitabine alone. Epigenetic synergy was also seen at endogenously hypermethylated tumor suppressor genes such as MLH1 and PDLIM4. Genome- wide studies showed that reactivation of hypermethylated genes by the combination was significantly better than that induced by decitabine alone or carboplatin alone. -

Rna-Sequencing Applications: Gene Expression Quantification and Methylator Phenotype Identification
The Texas Medical Center Library DigitalCommons@TMC The University of Texas MD Anderson Cancer Center UTHealth Graduate School of The University of Texas MD Anderson Cancer Biomedical Sciences Dissertations and Theses Center UTHealth Graduate School of (Open Access) Biomedical Sciences 8-2013 RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION Guoshuai Cai Follow this and additional works at: https://digitalcommons.library.tmc.edu/utgsbs_dissertations Part of the Bioinformatics Commons, Computational Biology Commons, and the Medicine and Health Sciences Commons Recommended Citation Cai, Guoshuai, "RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION" (2013). The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access). 386. https://digitalcommons.library.tmc.edu/utgsbs_dissertations/386 This Dissertation (PhD) is brought to you for free and open access by the The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences at DigitalCommons@TMC. It has been accepted for inclusion in The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences Dissertations and Theses (Open Access) by an authorized administrator of DigitalCommons@TMC. For more information, please contact [email protected]. RNA-SEQUENCING APPLICATIONS: GENE EXPRESSION QUANTIFICATION AND METHYLATOR PHENOTYPE IDENTIFICATION -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

The GALNT9, BNC1 and CCDC8 Genes Are Frequently Epigenetically Dysregulated in Breast Tumours That Metastasise to the Brain Rajendra P
Pangeni et al. Clinical Epigenetics (2015) 7:57 DOI 10.1186/s13148-015-0089-x RESEARCH Open Access The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain Rajendra P. Pangeni1, Prasanna Channathodiyil1, David S. Huen2, Lawrence W. Eagles1, Balraj K. Johal2, Dawar Pasha2, Natasa Hadjistephanou2, Oliver Nevell2, Claire L. Davies2, Ayobami I. Adewumi2, Hamida Khanom2, Ikroop S. Samra2, Vanessa C. Buzatto2, Preethi Chandrasekaran2, Thoraia Shinawi3, Timothy P. Dawson4, Katherine M. Ashton4, Charles Davis4, Andrew R. Brodbelt5, Michael D. Jenkinson5, Ivan Bièche6, Farida Latif3, John L. Darling1, Tracy J. Warr1 and Mark R. Morris1,2,3* Abstract Background: Tumour metastasis to the brain is a common and deadly development in certain cancers; 18–30 % of breast tumours metastasise to the brain. The contribution that gene silencing through epigenetic mechanisms plays in these metastatic tumours is not well understood. Results: We have carried out a bioinformatic screen of genome-wide breast tumour methylation data available at The Cancer Genome Atlas (TCGA) and a broad literature review to identify candidate genes that may contribute to breast to brain metastasis (BBM). This analysis identified 82 candidates. We investigated the methylation status of these genes using Combined Bisulfite and Restriction Analysis (CoBRA) and identified 21 genes frequently methylated in BBM. We have identified three genes, GALNT9, CCDC8 and BNC1, that were frequently methylated (55, 73 and 71 %, respectively) and silenced in BBM and infrequently methylated in primary breast tumours. CCDC8 was commonly methylated in brain metastases and their associated primary tumours whereas GALNT9 and BNC1 were methylated and silenced only in brain metastases, but not in the associated primary breast tumours from individual patients.