The GALNT9, BNC1 and CCDC8 Genes Are Frequently Epigenetically Dysregulated in Breast Tumours That Metastasise to the Brain Rajendra P
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Clinical Utility Gene Card For: 3-M Syndrome – Update 2013
European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156 & 2014 Macmillan Publishers Limited All rights reserved 1018-4813/14 www.nature.com/ejhg CLINICAL UTILITY GENE CARD UPDATE Clinical utility gene card for: 3-M syndrome – Update 2013 Muriel Holder-Espinasse*,1, Melita Irving1 and Vale´rie Cormier-Daire2 European Journal of Human Genetics (2014) 22, doi:10.1038/ejhg.2013.156; published online 31 July 2013 Update to: European Journal of Human Genetics (2011) 19, doi:10.1038/ejhg.2011.32; published online 2 March 2011 1. DISEASE CHARACTERISTICS nonsense and missense mutations c.4333C4T (p.Arg1445*) and 1.1 Name of the disease (synonyms) c.4391A4C (p.His1464Pro), respectively, render CUL7 deficient 3-M syndrome (gloomy face syndrome, dolichospondylic dysplasia). in recruiting ROC1, leading to impaired ubiquitination. OBSL1: microsatellites analysis of the locus (2q35-36.1) in con- 1.2 OMIM# of the disease sanguineous families. OBSL1: microsatellites analysis of the locus 273750. (2q35-36.1) in consanguineous families. Mutations induce non- sense mediated decay. Knockdown of OBSL1 in HEK293 cells 1.3 Name of the analysed genes or DNA/chromosome segments shows the role of this gene in the maintenance of normal levels of CUL7, OBSL1 and CCDC8.1–5 CUL7. Abnormal IGFBP2 andIGFBP5 mRNA levels in two patients with OBSL1 mutations, suggesting that OBSL1 modulates the 1.4 OMIM# of the gene(s) expression of IGFBP proteins. CCDC8: microsatellites analysis 609577 (CUL7), 610991 (OBSL1) and 614145 (CCDC8). at the locus (19q13.2-q13.32). CCDC8, 1-BP DUP, 612G and CCDC8, 1-BP. -

FBXW8 (NM 153348) Human Recombinant Protein Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP761689 FBXW8 (NM_153348) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Purified recombinant protein of Human F-box and WD repeat domain containing 8 (FBXW8), transcript variant 1, full length, with N-terminal HIS tag, expressed in E. coli, 50ug Species: Human Expression Host: E. coli Tag: N-His Predicted MW: 67.2 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 50mM Tris, pH8.0,8M Urea. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_699179 Locus ID: 26259 UniProt ID: Q8N3Y1 RefSeq Size: 4871 Cytogenetics: 12q24.22 RefSeq ORF: 1794 Synonyms: FBW6; FBW8; FBX29; FBXO29; FBXW6 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 FBXW8 (NM_153348) Human Recombinant Protein – TP761689 Summary: This gene encodes a member of the F-box protein family, members of which are characterized by an approximately 40 amino acid motif, the F-box. The F-box proteins constitute one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1- cullin-F-box), which function in phosphorylation-dependent ubiquitination. -

Identification and Validation of Methylation-Driven Genes Prognostic Signature for Recurrence of Laryngeal Squamous Cell Carcino
Cui et al. Cancer Cell Int (2020) 20:472 https://doi.org/10.1186/s12935-020-01567-3 Cancer Cell International PRIMARY RESEARCH Open Access Identifcation and validation of methylation-driven genes prognostic signature for recurrence of laryngeal squamous cell carcinoma by integrated bioinformatics analysis Jie Cui3†, Liping Wang2†, Waisheng Zhong4†, Zhen Chen5, Jie Chen4*, Hong Yang3* and Genglong Liu1* Abstract Background: Recurrence remains a major obstacle to long-term survival of laryngeal squamous cell carcinoma (LSCC). We conducted a genome-wide integrated analysis of methylation and the transcriptome to establish methyla- tion-driven genes prognostic signature (MDGPS) to precisely predict recurrence probability and optimize therapeutic strategies for LSCC. Methods: LSCC DNA methylation datasets and RNA sequencing (RNA-seq) dataset were acquired from the Cancer Genome Atlas (TCGA). MethylMix was applied to detect DNA methylation-driven genes (MDGs). By univariate and multivariate Cox regression analyses, fve genes of DNA MDGs was developed a recurrence-free survival (RFS)-related MDGPS. The predictive accuracy and clinical value of the MDGPS were evaluated by receiver operating characteristic (ROC) and decision curve analysis (DCA), and compared with TNM stage system. Additionally, prognostic value of MDGPS was validated by external Gene Expression Omnibus (GEO) database. According to 5 MDGs, the candidate small molecules for LSCC were screen out by the CMap database. To strengthen the bioinformatics analysis results, 30 pairs of clinical samples were evaluated by digoxigenin-labeled chromogenic in situ hybridization (CISH). Results: A total of 88 DNA MDGs were identifed, and fve RFS-related MDGs (LINC01354, CCDC8, PHYHD1, MAGEB2 and ZNF732) were chosen to construct a MDGPS. -

New PDF Document
888.267.4436 [email protected] www.origene.com Name:CLDN6 mouse monoclonal antibody, clone OTI3E3 (formerly 3E3) Catalog: TA507011 Product Data Sheet - TRUEMAB Components: • CLDN6 mouse monoclonal antibody, clone OTI3E3 (formerly 3E3) (TA507011) • WB positive control also available: 20ug myc-DDK tagged CLDN6 HEK293T over-expression lysate lyophilized in RIPA buffer (LC412034). (Reconstitute into 20ul of 1x SDS sample buffer before loading; load 5ul per lane as WB control or as desired) Amount: 100ul Immunogen: Full length human recombinant protein of human CLDN6(NP_067018) produced in HEK293T cell. Host: Mouse Isotype: IgG1 Species Reactivity: Human Guaranteed WB, IF Applications: Suggested WB 1:1000, IF 1:100, Dilutions: Concentration: 1 mg/ml Buffer: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Purification: Purified from mouse ascites fluids by affinity chromatography Storage Condition: Shipped at -20C or with ice packs. Upon delivery store at -20C. Dilute in PBS (pH7.3) if necessary. Stable for 12 months from date of receipt. Avoid repeated freeze-thaws. Target Target Name: Homo sapiens claudin 6 (CLDN6) Alternative Name: OTTHUMP00000159248; claudin 6 Database Link: NP_067018 Entrez Gene 9074 Human Function: Tight junctions represent one mode of cell-to-cell adhesion in epithelial or endothelial cell sheets, forming continuous seals around cells and serving as a physical barrier to prevent solutes and water from passing freely through the paracellular space. These junctions are comprised of sets of continuous networking strands in the outwardly facing cytoplasmic leaflet, with complementary grooves in the inwardly facing extracytoplasmic leaflet. This gene encodes a component of tight junction strands, which This product is to be used for laboratory only. -
Primepcr™Assay Validation Report
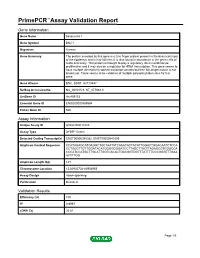
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

The DNA Methylation Landscape of Glioblastoma Disease Progression Shows Extensive Heterogeneity in Time and Space
bioRxiv preprint doi: https://doi.org/10.1101/173864; this version posted August 9, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space Johanna Klughammer1*, Barbara Kiesel2,3*, Thomas Roetzer3,4, Nikolaus Fortelny1, Amelie Kuchler1, Nathan C. Sheffield5, Paul Datlinger1, Nadine Peter3,4, Karl-Heinz Nenning6, Julia Furtner3,7, Martha Nowosielski8,9, Marco Augustin10, Mario Mischkulnig2,3, Thomas Ströbel3,4, Patrizia Moser11, Christian F. Freyschlag12, Jo- hannes Kerschbaumer12, Claudius Thomé12, Astrid E. Grams13, Günther Stockhammer8, Melitta Kitzwoegerer14, Stefan Oberndorfer15, Franz Marhold16, Serge Weis17, Johannes Trenkler18, Johanna Buchroithner19, Josef Pichler20, Johannes Haybaeck21,22, Stefanie Krassnig21, Kariem Madhy Ali23, Gord von Campe23, Franz Payer24, Camillo Sherif25, Julius Preiser26, Thomas Hauser27, Peter A. Winkler27, Waltraud Kleindienst28, Franz Würtz29, Tanisa Brandner-Kokalj29, Martin Stultschnig30, Stefan Schweiger31, Karin Dieckmann3,32, Matthias Preusser3,33, Georg Langs6, Bernhard Baumann10, Engelbert Knosp2,3, Georg Widhalm2,3, Christine Marosi3,33, Johannes A. Hainfellner3,4, Adelheid Woehrer3,4#§, Christoph Bock1,34,35# 1 CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 2 Department of Neurosurgery, Medical University of Vienna, Vienna, Austria. 3 Comprehensive Cancer Center, Central Nervous System Tumor Unit, Medical University of Vienna, Austria. 4 Institute of Neurology, Medical University of Vienna, Vienna, Austria. 5 Center for Public Health Genomics, University of Virginia, Charlottesville VA, USA. 6 Department of Biomedical Imaging and Image-guided Therapy, Computational Imaging Research Lab, Medical University of Vi- enna, Vienna, Austria. -

Identification of Novel DNA-Methylated Genes
Prostate Cancer and Prostatic Disease (2013) 16, 292–300 & 2013 Macmillan Publishers Limited All rights reserved 1365-7852/13 www.nature.com/pcan ORIGINAL ARTICLE Identification of novel DNA-methylated genes that correlate with human prostate cancer and high-grade prostatic intraepithelial neoplasia JM Devaney1, S Wang2,3, S Funda1, J Long2, DJ Taghipour2, R Tbaishat3, P Furbert-Harris2,4, M Ittmann5 and B Kwabi-Addo2,3 BACKGROUND: Prostate cancer (PCa) harbors a myriad of genomic and epigenetic defects. Cytosine methylation of CpG-rich promoter DNA is an important mechanism of epigenetic gene inactivation in PCa. There is considerable amount of data to suggest that DNA methylation-based biomarkers may be useful for the early detection and diagnosis of PCa. In addition, candidate gene- based studies have shown an association between specific gene methylation and alterations and clinicopathologic indicators of poor prognosis in PCa. METHODS: To more comprehensively identify DNA methylation alterations in PCa initiation and progression, we examined the methylation status of 485 577 CpG sites from regions with a broad spectrum of CpG densities, interrogating both gene-associated and non-associated regions using the recently developed Illumina 450K methylation platform. RESULTS: In all, we selected 33 promoter-associated novel CpG sites that were differentially methylated in high-grade prostatic intraepithelial neoplasia and PCa in comparison with benign prostate tissue samples (false discovery rate-adjusted P-value o0.05; b-value X0.2; fold change 41.5). Of the 33 genes, hierarchical clustering analysis demonstrated BNC1, FZD1, RPL39L, SYN2, LMX1B, CXXC5, ZNF783 and CYB5R2 as top candidate novel genes that are frequently methylated and whose methylation was associated with inactivation of gene expression in PCa cell lines. -

Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) Beyond SMARCA4 Mutations: a Comprehensive Genomic Analysis
cells Article Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT) beyond SMARCA4 Mutations: A Comprehensive Genomic Analysis Aurélie Auguste 1,Félix Blanc-Durand 2 , Marc Deloger 3 , Audrey Le Formal 1, Rohan Bareja 4,5, David C. Wilkes 4 , Catherine Richon 6,Béatrice Brunn 2, Olivier Caron 6, Mojgan Devouassoux-Shisheboran 7,Sébastien Gouy 2, Philippe Morice 2, Enrica Bentivegna 2, Andrea Sboner 4,5,8, Olivier Elemento 4,8, Mark A. Rubin 9 , Patricia Pautier 2, Catherine Genestie 10, Joanna Cyrta 4,9,11 and Alexandra Leary 1,2,* 1 Medical Oncologist, Gynecology Unit, Lead Translational Research Team, INSERM U981, Gustave Roussy, 94805 Villejuif, France; [email protected] (A.A.); [email protected] (A.L.F.) 2 Gynecological Unit, Department of Medicine, Gustave Roussy, 94805 Villejuif, France; [email protected] (F.B.-D.); [email protected] (B.B.); [email protected] (S.G.); [email protected] (P.M.); [email protected] (E.B.); [email protected] (P.P.) 3 Bioinformatics Core Facility, Gustave Roussy Cancer Center, UMS CNRS 3655/INSERM 23 AMMICA, 94805 Villejuif, France; [email protected] 4 Caryl and Israel Englander Institute for Precision Medicine, Weill Cornell Medicine, New York, NY 10001, USA; [email protected] (R.B.); [email protected] (D.C.W.); [email protected] (A.S.); [email protected] (O.E.); [email protected] (J.C.) 5 Institute for Computational Biomedicine, Weill Cornell -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

NIH Public Access Author Manuscript Mol Cell
NIH Public Access Author Manuscript Mol Cell. Author manuscript; available in PMC 2015 June 05. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Mol Cell. 2014 June 5; 54(5): 791–804. doi:10.1016/j.molcel.2014.03.047. The 3M complex maintains microtubule and genome integrity Jun Yan1, Feng Yan1, Zhijun Li1, Becky Sinnott4, Kathryn M. Cappell4,7, Yanbao Yu2, Jinyao Mo5, Joseph A. Duncan1,5, Xian Chen2, Valerie Cormier-Daire6, Angelique W. Whitehurst4,8, and Yue Xiong1,2,3,* 1Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295, USA. 2Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295, USA. 3Program in Molecular Biology and Biotechnology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295, USA. 4Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295, USA. 5Department of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599-7295, USA. 6University Paris Descartes, Department of Genetics and INSERM U781, Hospital Necker Enfants-Malades, Paris, France SUMMARY CUL7, OBSL1, and CCDC8 genes are mutated in a mutually exclusive manner in 3M and other growth retardation syndromes. The mechanism underlying the function of the three 3M genes in development is not known. We found that OBSL1 and CCDC8 form a complex with CUL7 and regulate the level and centrosomal localization of CUL7, respectively. CUL7 depletion results in altered microtubule dynamics, prometaphase arrest, tetraploidy and mitotic cell death. -

The Conserved DNMT1-Dependent Methylation Regions in Human Cells
Freeman et al. Epigenetics & Chromatin (2020) 13:17 https://doi.org/10.1186/s13072-020-00338-8 Epigenetics & Chromatin RESEARCH Open Access The conserved DNMT1-dependent methylation regions in human cells are vulnerable to neurotoxicant rotenone exposure Dana M. Freeman1 , Dan Lou1, Yanqiang Li1, Suzanne N. Martos1 and Zhibin Wang1,2,3* Abstract Background: Allele-specifc DNA methylation (ASM) describes genomic loci that maintain CpG methylation at only one inherited allele rather than having coordinated methylation across both alleles. The most prominent of these regions are germline ASMs (gASMs) that control the expression of imprinted genes in a parent of origin-dependent manner and are associated with disease. However, our recent report reveals numerous ASMs at non-imprinted genes. These non-germline ASMs are dependent on DNA methyltransferase 1 (DNMT1) and strikingly show the feature of random, switchable monoallelic methylation patterns in the mouse genome. The signifcance of these ASMs to human health has not been explored. Due to their shared allelicity with gASMs, herein, we propose that non-tradi- tional ASMs are sensitive to exposures in association with human disease. Results: We frst explore their conservancy in the human genome. Our data show that our putative non-germline ASMs were in conserved regions of the human genome and located adjacent to genes vital for neuronal develop- ment and maturation. We next tested the hypothesized vulnerability of these regions by exposing human embryonic kidney cell HEK293 with the neurotoxicant rotenone for 24 h. Indeed,14 genes adjacent to our identifed regions were diferentially expressed from RNA-sequencing. We analyzed the base-resolution methylation patterns of the predicted non-germline ASMs at two neurological genes, HCN2 and NEFM, with potential to increase the risk of neurodegenera- tion.