Rna-Sequencing Applications: Gene Expression Quantification and Methylator Phenotype Identification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Versican V2 Assembles the Extracellular Matrix Surrounding the Nodes of Ranvier in the CNS
The Journal of Neuroscience, June 17, 2009 • 29(24):7731–7742 • 7731 Cellular/Molecular Versican V2 Assembles the Extracellular Matrix Surrounding the Nodes of Ranvier in the CNS María T. Dours-Zimmermann,1 Konrad Maurer,2 Uwe Rauch,3 Wilhelm Stoffel,4 Reinhard Fa¨ssler,5 and Dieter R. Zimmermann1 Institutes of 1Surgical Pathology and 2Anesthesiology, University Hospital Zurich, CH-8091 Zurich, Switzerland, 3Vascular Wall Biology, Department of Experimental Medical Science, University of Lund, S-221 00 Lund, Sweden, 4Center for Biochemistry, Medical Faculty, University of Cologne, D-50931 Cologne, Germany, and 5Department of Molecular Medicine, Max Planck Institute of Biochemistry, D-82152 Martinsried, Germany The CNS-restricted versican splice-variant V2 is a large chondroitin sulfate proteoglycan incorporated in the extracellular matrix sur- rounding myelinated fibers and particularly accumulating at nodes of Ranvier. In vitro, it is a potent inhibitor of axonal growth and therefore considered to participate in the reduction of structural plasticity connected to myelination. To study the role of versican V2 during postnatal development, we designed a novel isoform-specific gene inactivation approach circumventing early embryonic lethality of the complete knock-out and preventing compensation by the remaining versican splice variants. These mice are viable and fertile; however, they display major molecular alterations at the nodes of Ranvier. While the clustering of nodal sodium channels and paranodal structures appear in versican V2-deficient mice unaffected, the formation of the extracellular matrix surrounding the nodes is largely impaired. The conjoint loss of tenascin-R and phosphacan from the perinodal matrix provide strong evidence that versican V2, possibly controlled by a nodal receptor, organizes the extracellular matrix assembly in vivo. -

Chromosomal Aberrations in Head and Neck Squamous Cell Carcinomas in Norwegian and Sudanese Populations by Array Comparative Genomic Hybridization
825-843 12/9/08 15:31 Page 825 ONCOLOGY REPORTS 20: 825-843, 2008 825 Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization ERIC ROMAN1,2, LEONARDO A. MEZA-ZEPEDA3, STINE H. KRESSE3, OLA MYKLEBOST3,4, ENDRE N. VASSTRAND2 and SALAH O. IBRAHIM1,2 1Department of Biomedicine, Faculty of Medicine and Dentistry, University of Bergen, Jonas Lies vei 91; 2Department of Oral Sciences - Periodontology, Faculty of Medicine and Dentistry, University of Bergen, Årstadveien 17, 5009 Bergen; 3Department of Tumor Biology, Institute for Cancer Research, Rikshospitalet-Radiumhospitalet Medical Center, Montebello, 0310 Oslo; 4Department of Molecular Biosciences, University of Oslo, Blindernveien 31, 0371 Oslo, Norway Received January 30, 2008; Accepted April 29, 2008 DOI: 10.3892/or_00000080 Abstract. We used microarray-based comparative genomic logical parameters showed little correlation, suggesting an hybridization to explore genome-wide profiles of chromosomal occurrence of gains/losses regardless of ethnic differences and aberrations in 26 samples of head and neck cancers compared clinicopathological status between the patients from the two to their pair-wise normal controls. The samples were obtained countries. Our findings indicate the existence of common from Sudanese (n=11) and Norwegian (n=15) patients. The gene-specific amplifications/deletions in these tumors, findings were correlated with clinicopathological variables. regardless of the source of the samples or attributed We identified the amplification of 41 common chromosomal carcinogenic risk factors. regions (harboring 149 candidate genes) and the deletion of 22 (28 candidate genes). Predominant chromosomal alterations Introduction that were observed included high-level amplification at 1q21 (harboring the S100A gene family) and 11q22 (including Head and neck squamous cell carcinoma (HNSCC), including several MMP family members). -

Keratins and Plakin Family Cytolinker Proteins Control the Length Of
RESEARCH ARTICLE Keratins and plakin family cytolinker proteins control the length of epithelial microridge protrusions Yasuko Inaba*, Vasudha Chauhan, Aaron Paul van Loon, Lamia Saiyara Choudhury, Alvaro Sagasti* Molecular, Cell and Developmental Biology Department and Molecular Biology Institute, University of California, Los Angeles, Los Angeles, United States Abstract Actin filaments and microtubules create diverse cellular protrusions, but intermediate filaments, the strongest and most stable cytoskeletal elements, are not known to directly participate in the formation of protrusions. Here we show that keratin intermediate filaments directly regulate the morphogenesis of microridges, elongated protrusions arranged in elaborate maze-like patterns on the surface of mucosal epithelial cells. We found that microridges on zebrafish skin cells contained both actin and keratin filaments. Keratin filaments stabilized microridges, and overexpressing keratins lengthened them. Envoplakin and periplakin, plakin family cytolinkers that bind F-actin and keratins, localized to microridges, and were required for their morphogenesis. Strikingly, plakin protein levels directly dictate microridge length. An actin-binding domain of periplakin was required to initiate microridge morphogenesis, whereas periplakin-keratin binding was required to elongate microridges. These findings separate microridge morphogenesis into distinct steps, expand our understanding of intermediate filament functions, and identify microridges as protrusions that integrate actin and intermediate filaments. *For correspondence: [email protected] (YI); Introduction [email protected] (AS) Cytoskeletal filaments are scaffolds for membrane protrusions that create a vast diversity of cell shapes. The three major classes of cytoskeletal elements—microtubules, actin filaments, and inter- Competing interests: The mediate filaments (IFs)—each have distinct mechanical and biochemical properties and associate authors declare that no with different regulatory proteins, suiting them to different functions. -

HHS Public Access Author Manuscript
HHS Public Access Author manuscript Author Manuscript Author ManuscriptAm J Med Author Manuscript Genet B Neuropsychiatr Author Manuscript Genet. Author manuscript; available in PMC 2015 December 01. Published in final edited form as: Am J Med Genet B Neuropsychiatr Genet. 2014 December ; 0(8): 673–683. doi:10.1002/ajmg.b.32272. Association and ancestry analysis of sequence variants in ADH and ALDH using alcohol-related phenotypes in a Native American community sample Qian Peng1,2,*, Ian R. Gizer3, Ondrej Libiger2, Chris Bizon4, Kirk C. Wilhelmsen4,5, Nicholas J. Schork1, and Cindy L. Ehlers6,* 1 Department of Human Biology, J. Craig Venter Institute, La Jolla, CA 92037 2 Scripps Translational Science Institute, The Scripps Research Institute, La Jolla, CA 92037 3 Department of Psychological Sciences, University of Missouri-Columbia, Columbia, MO 65211 4 Renaissance Computing Institute, University of North Carolina at Chapel Hill, Chapel Hill, NC 27517 5 Department of Genetics and Neurology, University of North Carolina at Chapel Hill, Chapel Hill, NC 27599 6 Department of Molecular and Cellular Neuroscience, The Scripps Research Institute, La Jolla, CA 92037 Abstract Higher rates of alcohol use and other drug-dependence have been observed in some Native American populations relative to other ethnic groups in the U.S. Previous studies have shown that alcohol dehydrogenase (ADH) genes and aldehyde dehydrogenase (ALDH) genes may affect the risk of development of alcohol dependence, and that polymorphisms within these genes may differentially affect risk for the disorder depending on the ethnic group evaluated. We evaluated variations in the ADH and ALDH genes in a large study investigating risk factors for substance use in a Native American population. -

Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research Through Collaboration October 15Th, 2019
The Gabriella Miller Kids First Pediatric Research Program (Kids First) Poster Session at ASHG Accelerating Pediatric Genomics Research through Collaboration October 15th, 2019 Background The Gabriella Miller Kids First Pediatric Research Program (Kids First) is a trans- NIH Common Fund program initiated in response to the 2014 Gabriella Miller Kids First Research Act. The program’s vision is to alleviate suffering from childhood cancer and structural birth defects by fostering collaborative research to uncover the etiology of these diseases and support data sharing within the pediatric research community. This is implemented through developing the Gabriella Miller Kids First Data Resource (Kids First Data Resource) and populating this resource with whole genome sequence datasets and associated clinical and phenotypic information. Both childhood cancers and structural birth defects are critical and costly conditions associated with substantial morbidity and mortality. Elucidating the underlying genetic etiology of these diseases has the potential to profoundly improve preventative measures, diagnostics, and therapeutic interventions. Purpose During this evening poster session, attendees will gain a broad understanding of the utility of the genomic data generated by Kids First, learn about the progress of Kids First X01 cohort projects, and observe demonstrations of the tools and functionalities of the recently launched Kids First Data Resource Portal. The session is an opportunity for the scientific community and public to engage with Kids First investigators, collaborators, and a growing community of researchers, patient foundations, and families. Several other NIH and external data efforts will present posters and be available to discuss collaboration opportunities as we work together to accelerate pediatric research. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

The Snakeskin-Mesh Complex of Smooth Septate Junction Restricts Yorkie to Regulate Intestinal Homeostasis in Drosophila
University of Massachusetts Medical School eScholarship@UMMS GSBS Dissertations and Theses Graduate School of Biomedical Sciences 2020-01-15 The Snakeskin-Mesh Complex of Smooth Septate Junction Restricts Yorkie to Regulate Intestinal Homeostasis in Drosophila Hsi-Ju Chen University of Massachusetts Medical School Let us know how access to this document benefits ou.y Follow this and additional works at: https://escholarship.umassmed.edu/gsbs_diss Part of the Cell Biology Commons, and the Genetics Commons Repository Citation Chen H. (2020). The Snakeskin-Mesh Complex of Smooth Septate Junction Restricts Yorkie to Regulate Intestinal Homeostasis in Drosophila. GSBS Dissertations and Theses. https://doi.org/10.13028/ 0r15-ze63. Retrieved from https://escholarship.umassmed.edu/gsbs_diss/1059 This material is brought to you by eScholarship@UMMS. It has been accepted for inclusion in GSBS Dissertations and Theses by an authorized administrator of eScholarship@UMMS. For more information, please contact [email protected]. The Snakeskin-Mesh Complex of Smooth Septate Junction Restricts Yorkie to Regulate Intestinal Homeostasis in Drosophila A Dissertation Presented by Hsi-Ju Chen Submitted to the Faculty of the University of Massachusetts Graduate School of Biomedical Sciences, Worcester In partial fulfilment of the requirement for the requirements for the Degree of Doctor of Philosophy November 19, 2019 TABLE OF CONTENTS ABSTRACT CHAPTER I..................................................................................................... -
Primepcr™Assay Validation Report
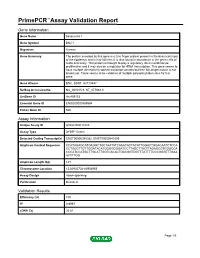
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

Age-Dependent Protein Abundance of Cytosolic Alcohol and Aldehyde Dehydrogenases in Human Liver S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2017/08/21/dmd.117.076463.DC2 http://dmd.aspetjournals.org/content/suppl/2017/06/12/dmd.117.076463.DC1 1521-009X/45/9/1044–1048$25.00 https://doi.org/10.1124/dmd.117.076463 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 45:1044–1048, September 2017 Copyright ª 2017 by The American Society for Pharmacology and Experimental Therapeutics Age-dependent Protein Abundance of Cytosolic Alcohol and Aldehyde Dehydrogenases in Human Liver s Deepak Kumar Bhatt, Andrea Gaedigk, Robin E. Pearce, J. Steven Leeder, and Bhagwat Prasad Department of Pharmaceutics, University of Washington, Seattle, Washington (D.K.B., B.P.); Department of Clinical Pharmacology, Toxicology & Therapeutic Innovation, Children’s Mercy-Kansas City, Missouri and School of Medicine, University of Missouri-Kansas City, Kansas City, Missouri (A.G., R.E.P., J.S.L.) Received April 21, 2017; accepted June 5, 2017 ABSTRACT Hepatic cytosolic alcohol and aldehyde dehydrogenases (ADHs and the adult levels, respectively. For all proteins, the abundance steeply ALDHs) catalyze the biotransformation of xenobiotics (e.g., cyclo- increased during the first year of life, which mostly reached adult Downloaded from phosphamide and ethanol) and vitamin A. Because age-dependent levels during early childhood (age between 1 and 6 years). Only for hepatic abundance of these proteins is unknown, we quantified ADH1A protein abundance in adults (age > 18 year) was ∼40% lower protein expression of ADHs and ALDH1A1 in a large cohort of pediatric relative to the early childhood group. Abundances of ADHs and and adult human livers by liquid chromatography coupled with tandem ALDH1A1 were not associated with sex in samples with age > 1 year mass spectrometry proteomics. -

A Single-Cell Transcriptomic Landscape of Primate Arterial Aging
ARTICLE https://doi.org/10.1038/s41467-020-15997-0 OPEN A single-cell transcriptomic landscape of primate arterial aging Weiqi Zhang 1,2,3,4,5,13, Shu Zhang6,7,13, Pengze Yan3,8,13, Jie Ren7,9,13, Moshi Song3,5,8, Jingyi Li2,3,8, Jinghui Lei4, Huize Pan2,3, Si Wang3,5,8, Xibo Ma3,10, Shuai Ma2,3,8, Hongyu Li2,3, Fei Sun2,3, Haifeng Wan3,5,11, ✉ ✉ ✉ Wei Li 3,5,11, Piu Chan4, Qi Zhou3,5,11, Guang-Hui Liu 2,3,4,5,8 , Fuchou Tang 6,7,9,12 & Jing Qu 3,5,11 Our understanding of how aging affects the cellular and molecular components of the vas- 1234567890():,; culature and contributes to cardiovascular diseases is still limited. Here we report a single-cell transcriptomic survey of aortas and coronary arteries in young and old cynomolgus monkeys. Our data define the molecular signatures of specialized arteries and identify eight markers discriminating aortic and coronary vasculatures. Gene network analyses characterize tran- scriptional landmarks that regulate vascular senility and position FOXO3A, a longevity- associated transcription factor, as a master regulator gene that is downregulated in six subtypes of monkey vascular cells during aging. Targeted inactivation of FOXO3A in human vascular endothelial cells recapitulates the major phenotypic defects observed in aged monkey arteries, verifying FOXO3A loss as a key driver for arterial endothelial aging. Our study provides a critical resource for understanding the principles underlying primate arterial aging and contributes important clues to future treatment of age-associated vascular disorders. 1 CAS Key Laboratory of Genomic and Precision Medicine, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing 100101, China. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -
Supplementary Table 2 Supplementary Table 1
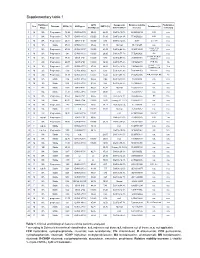
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a.