Introduction Summary of Contents: 1. Foundations of Thermodynamics 1.1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
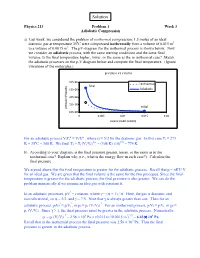
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

12/8 and 12/10/2010
PY105 C1 1. Help for Final exam has been posted on WebAssign. 2. The Final exam will be on Wednesday December 15th from 6-8 pm. First Law of Thermodynamics 3. You will take the exam in multiple rooms, divided as follows: SCI 107: Abbasi to Fasullo, as well as Khajah PHO 203: Flynn to Okuda, except for Khajah SCI B58: Ordonez to Zhang 1 2 Heat and Work done by a Gas Thermodynamics Initial: Consider a cylinder of ideal Thermodynamics is the study of systems involving gas at room temperature. Suppose the piston on top of energy in the form of heat and work. the cylinder is free to move vertically without friction. When the cylinder is placed in a container of hot water, heat Equilibrium: is transferred into the cylinder. Where does the heat energy go? Why does the volume increase? 3 4 The First Law of Thermodynamics The First Law of Thermodynamics The First Law is often written as: Some of the heat energy goes into raising the temperature of the gas (which is equivalent to raising the internal energy of the gas). The rest of it does work by raising the piston. ΔEQWint =− Conservation of energy leads to: QEW=Δ + int (the first law of thermodynamics) This form of the First Law says that the change in internal energy of a system is Q is the heat added to a system (or removed if it is negative) equal to the heat supplied to the system Eint is the internal energy of the system (the energy minus the work done by the system (usually associated with the motion of the atoms and/or molecules). -

17.Examples of the 1St Law Rev2.Nb
17. Examples of the First Law Introduction and Summary The First Law of Thermodynamics is basically a statement of Conservation of Energy. The total energy of a thermodynamic system is called the Internal Energy U. A system can have its Internal Energy changed (DU) in two major ways: (1) Heat Q > 0 can flow into the system from the surroundings, in which case the change in internal energy of the system increases: DU = Q. (2) The system can do work W > 0 on the surroundings, in which case the internal energy of the system decreases: DU = - W. Generally, the system interacts with the surroundings by exchanging both heat Q and doing work W, and the internal energy changes DU = Q - W This is the first law of thermodynamics and in it, W is the thermodynamic work done by the system on the surroundings. If the definition of mechanical work W were used instead of W, then the 1st Law would appear DU = Q + W because doing work on the system by the surroundings would increase the internal energy of the system DU = W. The 1st Law is simply the statement that the internal energy DU of a system can change in two ways: through heat Q absorption where, for example, the temperature changes, or by the system doing work -W, where a macroscopic parameter like volume changes. Below are some examples of using the 1st Law. The Work Done and the Heat Input During an Isobaric Expansion Suppose a gas is inside a cylinder that has a movable piston. The gas is in contact with a heat reservoir that is at a higher temperature than the gas so heat Q flows into the gas from the reservoir. -

BASIC THERMODYNAMICS REFERENCES: ENGINEERING THERMODYNAMICS by P.K.NAG 3RD EDITION LAWS of THERMODYNAMICS
Module I BASIC THERMODYNAMICS REFERENCES: ENGINEERING THERMODYNAMICS by P.K.NAG 3RD EDITION LAWS OF THERMODYNAMICS • 0 th law – when a body A is in thermal equilibrium with a body B, and also separately with a body C, then B and C will be in thermal equilibrium with each other. • Significance- measurement of property called temperature. A B C Evacuated tube 100o C Steam point Thermometric property 50o C (physical characteristics of reference body that changes with temperature) – rise of mercury in the evacuated tube 0o C bulb Steam at P =1ice atm T= 30oC REASONS FOR NOT TAKING ICE POINT AND STEAM POINT AS REFERENCE TEMPERATURES • Ice melts fast so there is a difficulty in maintaining equilibrium between pure ice and air saturated water. Pure ice Air saturated water • Extreme sensitiveness of steam point with pressure TRIPLE POINT OF WATER AS NEW REFERENCE TEMPERATURE • State at which ice liquid water and water vapor co-exist in equilibrium and is an easily reproducible state. This point is arbitrarily assigned a value 273.16 K • i.e. T in K = 273.16 X / Xtriple point • X- is any thermomertic property like P,V,R,rise of mercury, thermo emf etc. OTHER TYPES OF THERMOMETERS AND THERMOMETRIC PROPERTIES • Constant volume gas thermometers- pressure of the gas • Constant pressure gas thermometers- volume of the gas • Electrical resistance thermometer- resistance of the wire • Thermocouple- thermo emf CELCIUS AND KELVIN(ABSOLUTE) SCALE H Thermometer 2 Ar T in oC N Pg 2 O2 gas -273 oC (0 K) Absolute pressure P This absolute 0K cannot be obtatined (Pg+Patm) since it violates third law. -

3. the First Law of Thermodynamics and Related Definitions
09/30/08 1 3. THE FIRST LAW OF THERMODYNAMICS AND RELATED DEFINITIONS 3.1 General statement of the law Simply stated, the First Law states that the energy of the universe is constant. This is an empirical conservation principle (conservation of energy) and defines the term "energy." We will also see that "internal energy" is defined by the First Law. [As is always the case in atmospheric science, we will ignore the Einstein mass-energy equivalence relation.] Thus, the First Law states the following: 1. Heat is a form of energy. 2. Energy is conserved. The ways in which energy is transformed is of interest to us. The First Law is the second fundamental principle in (atmospheric) thermodynamics, and is used extensively. (The first was the equation of state.) One form of the First Law defines the relationship among work, internal energy, and heat input. In this chapter (and in subsequent chapters), we will explore many applications derived from the First Law and Equation of State. 3.2 Work of expansion If a system (parcel) is not in mechanical (pressure) equilibrium with its surroundings, it will expand or contract. Consider the example of a piston/cylinder system, in which the cylinder is filled with a gas. The cylinder undergoes an expansion or compression as shown below. Also shown in Fig. 3.1 is a p-V thermodynamic diagram, in which the physical state of the gas is represented by two thermodynamic variables: p,V in this case. [We will consider this and other types of thermodynamic diagrams in more detail later. -

Exercise 1 Q.5 State the Postulates of Kinetic Theory of Gases
Physics | 13.25 Solved Examples 10−3 × 250 N× 300 JEE Main/Boards = × 23 760 22400 6×× 10 273 Example 1: An electric bulb of volume 250cm³ was sealed off during manufacture at the pressure of −3 23 10× 250 ×× 6 10 × 273 15 10-3mm of Hg at 27°C. Find the number of air molecules N = = 8.02x10 mole 760×× 22400 300 in the bulb. NR Sol: PV= RT = N T = NkT AA Example 2: One gram-mole of oxygen at 27°C and one atmospheric pressure is enclosed in a vessel. Let N be the number of air molecules in the bulb. (a) Assuming the molecules to be moving with V , find V =250cm³, P = 10-3mm of Hg, rms 1 1 the number of collisions per second which molecules T1 = 273 + 27 = 300°K make with one square meter area of the vessel wall. As P11 V= Nk T where k is constant, then (b) The vessel is next thermally insulated and moved with a constant speed ν . It is then suddenly stopped. 10-3x 250 = N.k.300 … (i) 0 The process results in a rise of the temperature of the At N.T.P., one mole of air occupies a volume of 22.4 litre, gas by 1°C. Calculate the speed ν0 . 3 V00= 22400cm ,P= = 760mmofHg,760 mm of Hg, P 3N kT × 23 n = V = A T = 273° K and N0 = 6 10 molecules Sol: Formula based: & rms . Recall kT Mn ∴760 × 22400 = 6x1023 × k × 273 … (ii) the assumption of KTG. -

Industrial Process Industrial Process
INDUSTRIAL PROCESS INDUSTRIAL PROCESS SESSION 3 HEAT PROCESS sasanana1 [Pick the date] Session 3 Heat process Flash smelting Flash smelting (Finnish: Liekkisulatus) is a smelting process for sulfur-containing ores[1] including chalcopyrite. The process was developed by Outokumpu in Finland and first applied at the Harjavalta plant in 1949 for smelting copper ore.[2][3] It has also been adapted for nickel and lead production.[2] A second flash smelting system was developed by the International Nickel Company ('INCO') and has a different concentrate feed design compared to the Outokumpu flash furnace.[4] The Inco flash furnace has end-wall concentrate injection burners and a central waste gas off-take,[4] while the Outokumpu flash furnace has a water-cooled reaction shaft at one end of the vessel and a waste gas off-take at the other end.[5] While the INCO flash furnace at Sudbury was the first commercial use of oxygen flash smelting,[6] fewer smelters use the INCO flash furnace than the Outokumpu flash furnace.[4] Flash smelting with oxygen-enriched air (the 'reaction gas') makes use of the energy contained in the concentrate to supply most of the energy required by the furnaces.[4][5] The concentrate must be dried before it is injected into the furnaces and, in the case of the Outokumpu process, some of the furnaces use an optional heater to warm the reaction gas typically to 100–450 °C.[5] The reactions in the flash smelting furnaces produce copper matte, iron oxides and sulfur dioxide. The reacted particles fall into a bath at the bottom of the furnace, where the iron oxides react with fluxes, such as silica and limestone, to form a slag.[7] In most cases, the slag can be discarded, perhaps after some cleaning, and the matte is further treated in converters to produce blister copper. -

Thermodynamics
CHAPTER TWELVE THERMODYNAMICS 12.1 INTRODUCTION In previous chapter we have studied thermal properties of matter. In this chapter we shall study laws that govern thermal energy. We shall study the processes where work is 12.1 Introduction converted into heat and vice versa. In winter, when we rub 12.2 Thermal equilibrium our palms together, we feel warmer; here work done in rubbing 12.3 Zeroth law of produces the ‘heat’. Conversely, in a steam engine, the ‘heat’ Thermodynamics of the steam is used to do useful work in moving the pistons, 12.4 Heat, internal energy and which in turn rotate the wheels of the train. work In physics, we need to define the notions of heat, 12.5 First law of temperature, work, etc. more carefully. Historically, it took a thermodynamics long time to arrive at the proper concept of ‘heat’. Before the 12.6 Specific heat capacity modern picture, heat was regarded as a fine invisible fluid 12.7 Thermodynamic state filling in the pores of a substance. On contact between a hot variables and equation of body and a cold body, the fluid (called caloric) flowed from state the colder to the hotter body! This is similar to what happens 12.8 Thermodynamic processes when a horizontal pipe connects two tanks containing water 12.9 Heat engines up to different heights. The flow continues until the levels of 12.10 Refrigerators and heat water in the two tanks are the same. Likewise, in the ‘caloric’ pumps picture of heat, heat flows until the ‘caloric levels’ (i.e., the 12.11 Second law of temperatures) equalise. -

Lecture Outline
LECTURE OUTLINE 1. Ways of reaching saturation • Isobaric processes • Isobaric cooling (dew point temperature) • Isobaric and adiabatic cooling by evaporation of water (wet bulb temperature) Temperatura wilgotnego termometru) • Adiabatic and isobaric mixing 1 /26 WAYS OF REACHING SATURATION FORMATION AND DISSIPATION OF CLOUDS For simplicity, we assume that clouds form in the atmosphere when the water vapor reaches the saturation value, and f=100% (in reality this value should be greater than 100%). r f = v r p,T s ( ) An increase of relative humidity can be accomplished by: § increasing the amount of water vapor in the air (i.e. increasing rv); evaporation of water from a surface or via evaporation of rain falling through unsaturated air, § cooling of the air (decrease of rs(p,T)); isobaric cooling (e.g. radiative), adiabatic cooling of rising air, § mixing of two unsaturated parcels of air. 2 /26 THE COMBINED FIRST AND SECOND LAWS WILL BE APPLIED TO THE IDEALIZED THERMODYNAMIC REFERENCE PROCESSES ASSOCIATED WITH PHASE CHANGE OF WATER: 1. isobaric cooling 2. adiabatic isobaric processes 3. adiabatic and isobaric mixing 4. adiabatic expansion 3 /26 TEXTBOOKS • Salby, Chapter 5 • C&W, Chapter 4 • R&Y, Chapter 2 4 /26 ISOBARIC PROCESSES LEADING TO SATURATION THE AIR WITH WATER VAPOR 1. Isobaric cooling (p=const, rv=const) § Dew point temperature § Isobaric cooling with condensation (p=const, rt=const) 2. Isobaric and adiabatic cooling/heating by evaporation/condensation of water Izobaryczne i adiabatyczne ochładzanie poprzez odparowanie wody (p=const, a source of water vapor / water) § Wet bulb temperature § Equivalent temperature 3. Isobaric and adiabatic mixing (p=const, rt=const) 5 /26 ISOBARIC COOLING 6 /26 If vertical movement in the atmosphere is insignificant and a departure from the initial state is small, processes can be seen as isobaric processes. -

The First Law of Thermodynamics: Closed Systems Heat Transfer
The First Law of Thermodynamics: Closed Systems The first law of thermodynamics can be simply stated as follows: during an interaction between a system and its surroundings, the amount of energy gained by the system must be exactly equal to the amount of energy lost by the surroundings. A closed system can exchange energy with its surroundings through heat and work transfer. In other words, work and heat are the forms that energy can be transferred across the system boundary. Based on kinetic theory, heat is defined as the energy associated with the random motions of atoms and molecules. Heat Transfer Heat is defined as the form of energy that is transferred between two systems by virtue of a temperature difference. Note: there cannot be any heat transfer between two systems that are at the same temperature. Note: It is the thermal (internal) energy that can be stored in a system. Heat is a form of energy in transition and as a result can only be identified at the system boundary. Heat has energy units kJ (or BTU). Rate of heat transfer is the amount of heat transferred per unit time. Heat is a directional (or vector) quantity; thus, it has magnitude, direction and point of action. Notation: – Q (kJ) amount of heat transfer – Q° (kW) rate of heat transfer (power) – q (kJ/kg) ‐ heat transfer per unit mass – q° (kW/kg) ‐ power per unit mass Sign convention: Heat Transfer to a system is positive, and heat transfer from a system is negative. It means any heat transfer that increases the energy of a system is positive, and heat transfer that decreases the energy of a system is negative. -

Thermodynamic Functions at Isobaric Process of Van Der Waals Gases
Thermodynamic Functions at Isobaric Process of van der Waals Gases Akira Matsumoto Department of Material Sciences, College of Integrated Arts and Sciences, Osaka Prefecture University, Sakai, Osaka, 599-8531, Japan Reprint requests to Prof. A. Matsumoto; Fax. 81-0722-54-9927; E-mail: [email protected] Z. Naturforsch. 55 a, 851-855 (2000); received June 29, 2000 The thermodynamic functions for the van der Waals equation are investigated at isobaric process. The Gibbs free energy is expressed as the sum of the Helmholtz free energy and PV, and the volume in this case is described as the implicit function of the cubic equation for V in the van der Waals equation. Furthermore, the Gibbs free energy is given as a function of the reduced temperature, pressure and volume, introducing a reduced equation of state. Volume, enthalpy, entropy, heat capacity, thermal expansivity, and isothermal compressibility are given as functions of the reduced temperature, pressure and volume, respectively. Some thermodynamic quantities are calculated numerically and drawn graphically. The heat capacity, thermal expansivity, and isothermal compressibility diverge to infinity at the critical point. This suggests that a second-order phase transition may occur at the critical point. Key words: Van der Waals Gases; Isobaric Process; Second-order Phase Transition. 1. Introduction the properties of the determinant. In the case of these functions, a singularity will occur thermodynamically There is a continuing problem concerning the ther- at a critical point, while the volume V(T, P) becomes modynamic behaviours in a supercritical state. The a complicated expression as a root of the cubic equa- van der Waals equation as one of many empirical tion.