Second Law of Thermodynamics and Related Items
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
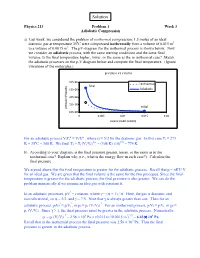
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Novel Hot Air Engine and Its Mathematical Model – Experimental Measurements and Numerical Analysis
POLLACK PERIODICA An International Journal for Engineering and Information Sciences DOI: 10.1556/606.2019.14.1.5 Vol. 14, No. 1, pp. 47–58 (2019) www.akademiai.com NOVEL HOT AIR ENGINE AND ITS MATHEMATICAL MODEL – EXPERIMENTAL MEASUREMENTS AND NUMERICAL ANALYSIS 1 Gyula KRAMER, 2 Gabor SZEPESI *, 3 Zoltán SIMÉNFALVI 1,2,3 Department of Chemical Machinery, Institute of Energy and Chemical Machinery University of Miskolc, Miskolc-Egyetemváros 3515, Hungary e-mail: [email protected], [email protected], [email protected] Received 11 December 2017; accepted 25 June 2018 Abstract: In the relevant literature there are many types of heat engines. One of those is the group of the so called hot air engines. This paper introduces their world, also introduces the new kind of machine that was developed and built at Department of Chemical Machinery, Institute of Energy and Chemical Machinery, University of Miskolc. Emphasizing the novelty of construction and the working principle are explained. Also the mathematical model of this new engine was prepared and compared to the real model of engine. Keywords: Hot, Air, Engine, Mathematical model 1. Introduction There are three types of volumetric heat engines: the internal combustion engines; steam engines; and hot air engines. The first one is well known, because it is on zenith nowadays. The steam machines are also well known, because their time has just passed, even the elder ones could see those in use. But the hot air engines are forgotten. Our aim is to consider that one. The history of hot air engines is 200 years old. -

Basic Thermodynamics-17ME33.Pdf
Module -1 Fundamental Concepts & Definitions & Work and Heat MODULE 1 Fundamental Concepts & Definitions Thermodynamics definition and scope, Microscopic and Macroscopic approaches. Some practical applications of engineering thermodynamic Systems, Characteristics of system boundary and control surface, examples. Thermodynamic properties; definition and units, intensive and extensive properties. Thermodynamic state, state point, state diagram, path and process, quasi-static process, cyclic and non-cyclic processes. Thermodynamic equilibrium; definition, mechanical equilibrium; diathermic wall, thermal equilibrium, chemical equilibrium, Zeroth law of thermodynamics, Temperature; concepts, scales, fixed points and measurements. Work and Heat Mechanics, definition of work and its limitations. Thermodynamic definition of work; examples, sign convention. Displacement work; as a part of a system boundary, as a whole of a system boundary, expressions for displacement work in various processes through p-v diagrams. Shaft work; Electrical work. Other types of work. Heat; definition, units and sign convention. 10 Hours 1st Hour Brain storming session on subject topics Thermodynamics definition and scope, Microscopic and Macroscopic approaches. Some practical applications of engineering thermodynamic Systems 2nd Hour Characteristics of system boundary and control surface, examples. Thermodynamic properties; definition and units, intensive and extensive properties. 3rd Hour Thermodynamic state, state point, state diagram, path and process, quasi-static -

Thermodynamics the Goal of Thermodynamics Is to Understand How Heat Can Be Converted to Work
Thermodynamics The goal of thermodynamics is to understand how heat can be converted to work Main lesson: Not all the heat energy can be converted to mechanical energy This is because heat energy comes with disorder (entropy), and overall disorder cannot decrease Temperature v T Random directions of velocity Higher temperature means higher velocities v Add up energy of all molecules: Internal Energy of gas U Mechanical energy: all atoms move in the same direction 1 Mv2 2 Statistical mechanics For one atom 1 1 1 1 1 1 3 E = mv2 + mv2 + mv2 = kT + kT + kT = kT h i h2 xi h2 yi h2 z i 2 2 2 2 Ideal gas: No Potential energy from attraction between atoms 3 U = NkT h i 2 v T Pressure v Pressure is caused because atoms T bounce off the wall Lx p − x px ∆px =2px 2L ∆t = x vx ∆p 2mv2 mv2 F = x = x = x ∆t 2Lx Lx ∆p 2mv2 mv2 F = x = x = x ∆t 2Lx Lx 1 mv2 =2 kT = kT h xi ⇥ 2 kT F = Lx Pressure v F kT 1 kT A = LyLz P = = = A Lx LyLz V NkT Many particles P = V Lx PV = NkT Volume V = LxLyLz Work Lx ∆V = A ∆Lx v A = LyLz Work done BY the gas ∆W = F ∆Lx We can write this as ∆W =(PA) ∆Lx = P ∆V This is useful because the body could have a generic shape Internal energy of gas decreases U U ∆W ! − Gas expands, work is done BY the gas P dW = P dV Work done is Area under curve V P Gas is pushed in, work is done ON the gas dW = P dV − Work done is negative of Area under curve V By convention, we use POSITIVE sign for work done BY the gas Getting work from Heat Gas expands, work is done BY the gas P dW = P dV V Volume in increases Internal energy decreases .. -

Thermodynamic Entropy As an Indicator for Urban Sustainability?
Available online at www.sciencedirect.com ScienceDirect Procedia Engineering 00 (2017) 000–000 www.elsevier.com/locate/procedia Urban Transitions Conference, Shanghai, September 2016 Thermodynamic entropy as an indicator for urban sustainability? Ben Purvisa,*, Yong Maoa, Darren Robinsona aLaboratory of Urban Complexity and Sustainability, University of Nottingham, NG7 2RD, UK bSecond affiliation, Address, City and Postcode, Country Abstract As foci of economic activity, resource consumption, and the production of material waste and pollution, cities represent both a major hurdle and yet also a source of great potential for achieving the goal of sustainability. Motivated by the desire to better understand and measure sustainability in quantitative terms we explore the applicability of thermodynamic entropy to urban systems as a tool for evaluating sustainability. Having comprehensively reviewed the application of thermodynamic entropy to urban systems we argue that the role it can hope to play in characterising sustainability is limited. We show that thermodynamic entropy may be considered as a measure of energy efficiency, but must be complimented by other indices to form part of a broader measure of urban sustainability. © 2017 The Authors. Published by Elsevier Ltd. Peer-review under responsibility of the organizing committee of the Urban Transitions Conference. Keywords: entropy; sustainability; thermodynamics; city; indicators; exergy; second law 1. Introduction The notion of using thermodynamic concepts as a tool for better understanding the problems relating to “sustainability” is not a new one. Ayres and Kneese (1969) [1] are credited with popularising the use of physical conservation principles in economic thinking. Georgescu-Roegen was the first to consider the relationship between the second law of thermodynamics and the degradation of natural resources [2]. -

25 Kw Low-Temperature Stirling Engine for Heat Recovery, Solar, and Biomass Applications
25 kW Low-Temperature Stirling Engine for Heat Recovery, Solar, and Biomass Applications Lee SMITHa, Brian NUELa, Samuel P WEAVERa,*, Stefan BERKOWERa, Samuel C WEAVERb, Bill GROSSc aCool Energy, Inc, 5541 Central Avenue, Boulder CO 80301 bProton Power, Inc, 487 Sam Rayburn Parkway, Lenoir City TN 37771 cIdealab, 130 W. Union St, Pasadena CA 91103 *Corresponding author: [email protected] Keywords: Stirling engine, waste heat recovery, concentrating solar power, biomass power generation, low-temperature power generation, distributed generation ABSTRACT This paper covers the design, performance optimization, build, and test of a 25 kW Stirling engine that has demonstrated > 60% of the Carnot limit for thermal to electrical conversion efficiency at test conditions of 329 °C hot side temperature and 19 °C rejection temperature. These results were enabled by an engine design and construction that has minimal pressure drop in the gas flow path, thermal conduction losses that are limited by design, and which employs a novel rotary drive mechanism. Features of this engine design include high-surface- area heat exchangers, nitrogen as the working fluid, a single-acting alpha configuration, and a design target for operation between 150 °C and 400 °C. 1 1. INTRODUCTION Since 2006, Cool Energy, Inc. (CEI) has designed, fabricated, and tested five generations of low-temperature (150 °C to 400 °C) Stirling engines that drive internally integrated electric alternators. The fifth generation of engine built by Cool Energy is rated at 25 kW of electrical power output, and is trade-named the ThermoHeart® Engine. Sources of low-to-medium temperature thermal energy, such as internal combustion engine exhaust, industrial waste heat, flared gas, and small-scale solar heat, have relatively few methods available for conversion into more valuable electrical energy, and the thermal energy is usually wasted. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

The Concept of Irreversibility: Its Use in the Sustainable Development and Precautionary Principle Literatures
The Electronic Journal of Sustainable Development (2007) 1(1) The concept of irreversibility: its use in the sustainable development and precautionary principle literatures Dr. Neil A. Manson* *Dr. Neil A. Manson is an assistant professor of philosophy at the University of Mississippi. Email: namanson =a= olemiss.edu (replace =a= with @) Writers on sustainable development and the Precautionary Principle frequently invoke the concept of irreversibil- ity. This paper gives a detailed analysis of that concept. Three senses of “irreversible” are distinguished: thermody- namic, medical, and economic. For each sense, an ontology (a realm of application for the term) and a normative status (whether the term is purely descriptive or partially evaluative) are identified. Then specific uses of “irreversible” in the literatures on sustainable development and the Precautionary Principle are analysed. The paper concludes with some advice on how “irreversible” should be used in the context of environmental decision-making. Key Words irreversibility; sustainability; sustainable development; the Precautionary Principle; cost-benefit analysis; thermody- namics; medicine; economics; restoration I. Introduction “idea people” as a political slogan (notice the alliteration in “compassionate conservatism”) with the content to be The term “compassionate conservatism” entered public provided later. The whole point of a political slogan is to consciousness during the 2000 U.S. presidential cam- tap into some vaguely held sentiment. Political slogans paign. It was created in response to the charge (made are not meant to be sharply defined, for sharp defini- frequently during the Reagan era, and repeated after the tions draw sharp boundaries and sharp boundaries force Republican takeover of Congress in 1994) that Repub- undecided voters out of one’s constituency. -

Supercritical CO 2 Cycles for Gas Turbine
Power Gen International December 8-10, Las Vegas, Nevada SUPERCRITICAL CO2 CYCLES FOR GAS TURBINE COMBINED CYCLE POWER PLANTS Timothy J. Held Chief Technology Officer Echogen Power Systems, LLC Akron, Ohio [email protected] ABSTRACT Supercritical carbon dioxide (sCO2) used as the working fluid in closed loop power conversion cycles offers significant advantages over steam and organic fluid based Rankine cycles. Echogen Power Systems LLC has developed several variants of sCO2 cycles that are optimized for bottoming and heat recovery applications. In contrast to cycles used in previous nuclear and CSP studies, these cycles are highly effective in extracting heat from a sensible thermal source such as gas turbine exhaust or industrial process waste heat, and then converting it to power. In this study, conceptual designs of sCO2 heat recovery systems are developed for gas turbine combined cycle (GTCC) power generation over a broad range of system sizes, ranging from distributed generation (~5MW) to utility scale (> 500MW). Advanced cycle simulation tools employing non-linear multivariate constrained optimization processes are combined with system and plant cost models to generate families of designs with different cycle topologies. The recently introduced EPS100 [1], the first commercial-scale sCO2 heat recovery engine, is used to validate the results of the cost and performance models. The results of the simulation process are shown as system installed cost as a function of power, which allows objective comparisons between different cycle architectures, and to other power generation technologies. Comparable system cost and performance studies for conventional steam-based GTCC are presented on the basis of GT-Pro™ simulations [2]. -

The Influence of Thermodynamic Ideas on Ecological Economics: an Interdisciplinary Critique
Sustainability 2009, 1, 1195-1225; doi:10.3390/su1041195 OPEN ACCESS sustainability ISSN 2071-1050 www.mdpi.com/journal/sustainability Article The Influence of Thermodynamic Ideas on Ecological Economics: An Interdisciplinary Critique Geoffrey P. Hammond 1,2,* and Adrian B. Winnett 1,3 1 Institute for Sustainable Energy & the Environment (I•SEE), University of Bath, Bath, BA2 7AY, UK 2 Department of Mechanical Engineering, University of Bath, Bath, BA2 7AY, UK 3 Department of Economics, University of Bath, Bath, BA2 7AY, UK; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +44-12-2538-6168; Fax: +44-12-2538-6928. Received: 10 October 2009 / Accepted: 24 November 2009 / Published: 1 December 2009 Abstract: The influence of thermodynamics on the emerging transdisciplinary field of ‗ecological economics‘ is critically reviewed from an interdisciplinary perspective. It is viewed through the lens provided by the ‗bioeconomist‘ Nicholas Georgescu-Roegen (1906–1994) and his advocacy of ‗the Entropy Law‘ as a determinant of economic scarcity. It is argued that exergy is a more easily understood thermodynamic property than is entropy to represent irreversibilities in complex systems, and that the behaviour of energy and matter are not equally mirrored by thermodynamic laws. Thermodynamic insights as typically employed in ecological economics are simply analogues or metaphors of reality. They should therefore be empirically tested against the real world. Keywords: thermodynamic analysis; energy; entropy; exergy; ecological economics; environmental economics; exergoeconomics; complexity; natural capital; sustainability Sustainability 2009, 1 1196 ―A theory is the more impressive, the greater the simplicity of its premises is, the more different kinds of things it relates, and the more extended is its area of applicability. -

Removing the Mystery of Entropy and Thermodynamics – Part II Harvey S
Table IV. Example of audit of energy loss in collision (with a mass attached to the end of the spring). Removing the Mystery of Entropy and Thermodynamics – Part II Harvey S. Leff, Reed College, Portland, ORa,b Part II of this five-part series is focused on further clarification of entropy and thermodynamics. We emphasize that entropy is a state function with a numerical value for any substance in thermodynamic equilibrium with its surroundings. The inter- pretation of entropy as a “spreading function” is suggested by the Clausius algorithm. The Mayer-Joule principle is shown to be helpful in understanding entropy changes for pure work processes. Furthermore, the entropy change when a gas expands or is compressed, and when two gases are mixed, can be understood qualitatively in terms of spatial energy spreading. The question- answer format of Part I1 is continued, enumerating main results in Key Points 2.1-2.6. • What is the significance of entropy being a state Although the linear correlation in Fig. 1 is quite striking, the function? two points to be emphasized here are: (i) the entropy value In Part I, we showed that the entropy of a room temperature of each solid is dependent on the energy added to and stored solid can be calculated at standard temperature and pressure by it, and (ii) the amount of energy needed for the heating using heat capacity data from near absolute zero, denoted by T process from T = 0 + K to T = 298.15 K differs from solid to = 0+, to room temperature, Tf = 298.15 K.