Chbe 11: Chemical Engineering Thermodynamics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
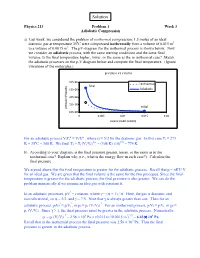
3-1 Adiabatic Compression
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Thermodynamics of Ideal Gases
D Thermodynamics of ideal gases An ideal gas is a nice “laboratory” for understanding the thermodynamics of a fluid with a non-trivial equation of state. In this section we shall recapitulate the conventional thermodynamics of an ideal gas with constant heat capacity. For more extensive treatments, see for example [67, 66]. D.1 Internal energy In section 4.1 we analyzed Bernoulli’s model of a gas consisting of essentially 1 2 non-interacting point-like molecules, and found the pressure p = 3 ½ v where v is the root-mean-square average molecular speed. Using the ideal gas law (4-26) the total molecular kinetic energy contained in an amount M = ½V of the gas becomes, 1 3 3 Mv2 = pV = nRT ; (D-1) 2 2 2 where n = M=Mmol is the number of moles in the gas. The derivation in section 4.1 shows that the factor 3 stems from the three independent translational degrees of freedom available to point-like molecules. The above formula thus expresses 1 that in a mole of a gas there is an internal kinetic energy 2 RT associated with each translational degree of freedom of the point-like molecules. Whereas monatomic gases like argon have spherical molecules and thus only the three translational degrees of freedom, diatomic gases like nitrogen and oxy- Copyright °c 1998{2004, Benny Lautrup Revision 7.7, January 22, 2004 662 D. THERMODYNAMICS OF IDEAL GASES gen have stick-like molecules with two extra rotational degrees of freedom or- thogonally to the bridge connecting the atoms, and multiatomic gases like carbon dioxide and methane have the three extra rotational degrees of freedom. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

12/8 and 12/10/2010
PY105 C1 1. Help for Final exam has been posted on WebAssign. 2. The Final exam will be on Wednesday December 15th from 6-8 pm. First Law of Thermodynamics 3. You will take the exam in multiple rooms, divided as follows: SCI 107: Abbasi to Fasullo, as well as Khajah PHO 203: Flynn to Okuda, except for Khajah SCI B58: Ordonez to Zhang 1 2 Heat and Work done by a Gas Thermodynamics Initial: Consider a cylinder of ideal Thermodynamics is the study of systems involving gas at room temperature. Suppose the piston on top of energy in the form of heat and work. the cylinder is free to move vertically without friction. When the cylinder is placed in a container of hot water, heat Equilibrium: is transferred into the cylinder. Where does the heat energy go? Why does the volume increase? 3 4 The First Law of Thermodynamics The First Law of Thermodynamics The First Law is often written as: Some of the heat energy goes into raising the temperature of the gas (which is equivalent to raising the internal energy of the gas). The rest of it does work by raising the piston. ΔEQWint =− Conservation of energy leads to: QEW=Δ + int (the first law of thermodynamics) This form of the First Law says that the change in internal energy of a system is Q is the heat added to a system (or removed if it is negative) equal to the heat supplied to the system Eint is the internal energy of the system (the energy minus the work done by the system (usually associated with the motion of the atoms and/or molecules). -

Chem 322, Physical Chemistry II: Lecture Notes
Chem 322, Physical Chemistry II: Lecture notes M. Kuno December 20, 2012 2 Contents 1 Preface 1 2 Revision history 3 3 Introduction 5 4 Relevant Reading in McQuarrie 9 5 Summary of common equations 11 6 Summary of common symbols 13 7 Units 15 8 Math interlude 21 9 Thermo definitions 35 10 Equation of state: Ideal gases 37 11 Equation of state: Real gases 47 12 Equation of state: Condensed phases 55 13 First law of thermodynamics 59 14 Heat capacities, internal energy and enthalpy 79 15 Thermochemistry 111 16 Entropy and the 2nd and 3rd Laws of Thermodynamics 123 i ii CONTENTS 17 Free energy (Helmholtz and Gibbs) 159 18 The fundamental equations of thermodynamics 179 19 Application of Free Energy to Phase Transitions 187 20 Mixtures 197 21 Equilibrium 223 22 Recap 257 23 Kinetics 263 Chapter 1 Preface What is the first law of thermodynamics? You do not talk about thermodynamics What is the second law of thermodynamics? You do not talk about thermodynamics 1 2 CHAPTER 1. PREFACE Chapter 2 Revision history 2/05: Original build 11/07: Corrected all typos caught to date and added some more material 1/08: Added more stuff and demos. Made the equilibrium section more pedagogical. 5/08: Caught more typos and made appropriate fixes to some nomenclature mistakes. 12/11: Started the next round of text changes and error corrections since I’m teaching this class again in spring 2012. Woah! I caught a whole bunch of problems that were present in the last version. It makes you wonder what I was thinking. -

17.Examples of the 1St Law Rev2.Nb
17. Examples of the First Law Introduction and Summary The First Law of Thermodynamics is basically a statement of Conservation of Energy. The total energy of a thermodynamic system is called the Internal Energy U. A system can have its Internal Energy changed (DU) in two major ways: (1) Heat Q > 0 can flow into the system from the surroundings, in which case the change in internal energy of the system increases: DU = Q. (2) The system can do work W > 0 on the surroundings, in which case the internal energy of the system decreases: DU = - W. Generally, the system interacts with the surroundings by exchanging both heat Q and doing work W, and the internal energy changes DU = Q - W This is the first law of thermodynamics and in it, W is the thermodynamic work done by the system on the surroundings. If the definition of mechanical work W were used instead of W, then the 1st Law would appear DU = Q + W because doing work on the system by the surroundings would increase the internal energy of the system DU = W. The 1st Law is simply the statement that the internal energy DU of a system can change in two ways: through heat Q absorption where, for example, the temperature changes, or by the system doing work -W, where a macroscopic parameter like volume changes. Below are some examples of using the 1st Law. The Work Done and the Heat Input During an Isobaric Expansion Suppose a gas is inside a cylinder that has a movable piston. The gas is in contact with a heat reservoir that is at a higher temperature than the gas so heat Q flows into the gas from the reservoir. -

Physical Chemistry I
Physical Chemistry I Andrew Rosen August 19, 2013 Contents 1 Thermodynamics 5 1.1 Thermodynamic Systems and Properties . .5 1.1.1 Systems vs. Surroundings . .5 1.1.2 Types of Walls . .5 1.1.3 Equilibrium . .5 1.1.4 Thermodynamic Properties . .5 1.2 Temperature . .6 1.3 The Mole . .6 1.4 Ideal Gases . .6 1.4.1 Boyle’s and Charles’ Laws . .6 1.4.2 Ideal Gas Equation . .7 1.5 Equations of State . .7 2 The First Law of Thermodynamics 7 2.1 Classical Mechanics . .7 2.2 P-V Work . .8 2.3 Heat and The First Law of Thermodynamics . .8 2.4 Enthalpy and Heat Capacity . .9 2.5 The Joule and Joule-Thomson Experiments . .9 2.6 The Perfect Gas . 10 2.7 How to Find Pressure-Volume Work . 10 2.7.1 Non-Ideal Gas (Van der Waals Gas) . 10 2.7.2 Ideal Gas . 10 2.7.3 Reversible Adiabatic Process in a Perfect Gas . 11 2.8 Summary of Calculating First Law Quantities . 11 2.8.1 Constant Pressure (Isobaric) Heating . 11 2.8.2 Constant Volume (Isochoric) Heating . 11 2.8.3 Reversible Isothermal Process in a Perfect Gas . 12 2.8.4 Reversible Adiabatic Process in a Perfect Gas . 12 2.8.5 Adiabatic Expansion of a Perfect Gas into a Vacuum . 12 2.8.6 Reversible Phase Change at Constant T and P ...................... 12 2.9 Molecular Modes of Energy Storage . 12 2.9.1 Degrees of Freedom . 12 1 2.9.2 Classical Mechanics . -

BASIC THERMODYNAMICS REFERENCES: ENGINEERING THERMODYNAMICS by P.K.NAG 3RD EDITION LAWS of THERMODYNAMICS
Module I BASIC THERMODYNAMICS REFERENCES: ENGINEERING THERMODYNAMICS by P.K.NAG 3RD EDITION LAWS OF THERMODYNAMICS • 0 th law – when a body A is in thermal equilibrium with a body B, and also separately with a body C, then B and C will be in thermal equilibrium with each other. • Significance- measurement of property called temperature. A B C Evacuated tube 100o C Steam point Thermometric property 50o C (physical characteristics of reference body that changes with temperature) – rise of mercury in the evacuated tube 0o C bulb Steam at P =1ice atm T= 30oC REASONS FOR NOT TAKING ICE POINT AND STEAM POINT AS REFERENCE TEMPERATURES • Ice melts fast so there is a difficulty in maintaining equilibrium between pure ice and air saturated water. Pure ice Air saturated water • Extreme sensitiveness of steam point with pressure TRIPLE POINT OF WATER AS NEW REFERENCE TEMPERATURE • State at which ice liquid water and water vapor co-exist in equilibrium and is an easily reproducible state. This point is arbitrarily assigned a value 273.16 K • i.e. T in K = 273.16 X / Xtriple point • X- is any thermomertic property like P,V,R,rise of mercury, thermo emf etc. OTHER TYPES OF THERMOMETERS AND THERMOMETRIC PROPERTIES • Constant volume gas thermometers- pressure of the gas • Constant pressure gas thermometers- volume of the gas • Electrical resistance thermometer- resistance of the wire • Thermocouple- thermo emf CELCIUS AND KELVIN(ABSOLUTE) SCALE H Thermometer 2 Ar T in oC N Pg 2 O2 gas -273 oC (0 K) Absolute pressure P This absolute 0K cannot be obtatined (Pg+Patm) since it violates third law. -

3. the First Law of Thermodynamics and Related Definitions
09/30/08 1 3. THE FIRST LAW OF THERMODYNAMICS AND RELATED DEFINITIONS 3.1 General statement of the law Simply stated, the First Law states that the energy of the universe is constant. This is an empirical conservation principle (conservation of energy) and defines the term "energy." We will also see that "internal energy" is defined by the First Law. [As is always the case in atmospheric science, we will ignore the Einstein mass-energy equivalence relation.] Thus, the First Law states the following: 1. Heat is a form of energy. 2. Energy is conserved. The ways in which energy is transformed is of interest to us. The First Law is the second fundamental principle in (atmospheric) thermodynamics, and is used extensively. (The first was the equation of state.) One form of the First Law defines the relationship among work, internal energy, and heat input. In this chapter (and in subsequent chapters), we will explore many applications derived from the First Law and Equation of State. 3.2 Work of expansion If a system (parcel) is not in mechanical (pressure) equilibrium with its surroundings, it will expand or contract. Consider the example of a piston/cylinder system, in which the cylinder is filled with a gas. The cylinder undergoes an expansion or compression as shown below. Also shown in Fig. 3.1 is a p-V thermodynamic diagram, in which the physical state of the gas is represented by two thermodynamic variables: p,V in this case. [We will consider this and other types of thermodynamic diagrams in more detail later. -

Exercise 1 Q.5 State the Postulates of Kinetic Theory of Gases
Physics | 13.25 Solved Examples 10−3 × 250 N× 300 JEE Main/Boards = × 23 760 22400 6×× 10 273 Example 1: An electric bulb of volume 250cm³ was sealed off during manufacture at the pressure of −3 23 10× 250 ×× 6 10 × 273 15 10-3mm of Hg at 27°C. Find the number of air molecules N = = 8.02x10 mole 760×× 22400 300 in the bulb. NR Sol: PV= RT = N T = NkT AA Example 2: One gram-mole of oxygen at 27°C and one atmospheric pressure is enclosed in a vessel. Let N be the number of air molecules in the bulb. (a) Assuming the molecules to be moving with V , find V =250cm³, P = 10-3mm of Hg, rms 1 1 the number of collisions per second which molecules T1 = 273 + 27 = 300°K make with one square meter area of the vessel wall. As P11 V= Nk T where k is constant, then (b) The vessel is next thermally insulated and moved with a constant speed ν . It is then suddenly stopped. 10-3x 250 = N.k.300 … (i) 0 The process results in a rise of the temperature of the At N.T.P., one mole of air occupies a volume of 22.4 litre, gas by 1°C. Calculate the speed ν0 . 3 V00= 22400cm ,P= = 760mmofHg,760 mm of Hg, P 3N kT × 23 n = V = A T = 273° K and N0 = 6 10 molecules Sol: Formula based: & rms . Recall kT Mn ∴760 × 22400 = 6x1023 × k × 273 … (ii) the assumption of KTG. -

Industrial Process Industrial Process
INDUSTRIAL PROCESS INDUSTRIAL PROCESS SESSION 3 HEAT PROCESS sasanana1 [Pick the date] Session 3 Heat process Flash smelting Flash smelting (Finnish: Liekkisulatus) is a smelting process for sulfur-containing ores[1] including chalcopyrite. The process was developed by Outokumpu in Finland and first applied at the Harjavalta plant in 1949 for smelting copper ore.[2][3] It has also been adapted for nickel and lead production.[2] A second flash smelting system was developed by the International Nickel Company ('INCO') and has a different concentrate feed design compared to the Outokumpu flash furnace.[4] The Inco flash furnace has end-wall concentrate injection burners and a central waste gas off-take,[4] while the Outokumpu flash furnace has a water-cooled reaction shaft at one end of the vessel and a waste gas off-take at the other end.[5] While the INCO flash furnace at Sudbury was the first commercial use of oxygen flash smelting,[6] fewer smelters use the INCO flash furnace than the Outokumpu flash furnace.[4] Flash smelting with oxygen-enriched air (the 'reaction gas') makes use of the energy contained in the concentrate to supply most of the energy required by the furnaces.[4][5] The concentrate must be dried before it is injected into the furnaces and, in the case of the Outokumpu process, some of the furnaces use an optional heater to warm the reaction gas typically to 100–450 °C.[5] The reactions in the flash smelting furnaces produce copper matte, iron oxides and sulfur dioxide. The reacted particles fall into a bath at the bottom of the furnace, where the iron oxides react with fluxes, such as silica and limestone, to form a slag.[7] In most cases, the slag can be discarded, perhaps after some cleaning, and the matte is further treated in converters to produce blister copper. -

Thermodynamics
...Thermodynamics Clausius Inequality R dq Existence of a definite integral T Entropy - a state variable Second Law stated as principle of increasing entropy Entropy changes in system and the surroundings October 9, 2018 1 / 20 The Principle of Increasing Entropy Isolated system, or System plus Surroundings enclosed by an adiabatic wall, or entire universe. d¯q = 0 ) ∆S ≥ 0. The entropy of a thermally isolated ensemble can never decrease. This defines a direction of time. The Second Law is the ONLY fundamental equation in physics which violates CPT Symmetry. Boltzmann argued that its validity was not based on the inherent laws of nature but rather on the choice of extremely improbable initial conditions. October 9, 2018 2 / 20 The Central Equation of Thermodynamics Combine the first and second laws. dU = d¯Q + d¯W ! dU = TdS - PdV Derived for infinitesimal reversible processes in differential form. This equation involves only state variables. Can be integrated to relate changes of state variables independent of the path of integration. Integration of equation can be applied to irreversible processes. Can be written TdS = dU + PdV , relating entropy to more easily measurable quantities. U and V are extensive quantities, so S must also be extensive. October 9, 2018 3 / 20 Caveat: PdV implies mechanical work compressing a fluid. In general d¯W should include all forms of work. e.g. For electric charge (Z) and emf E, dU = TdS − PdV + EdZ. For a rubber band length L under tension F, dU = TdS + FdL. October 9, 2018 4 / 20 Entropy of an ideal gas For an Ideal gas U = U(T ), so dU = Cv dT .