3. the First Law of Thermodynamics and Related Definitions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
3-1 Adiabatic Compression
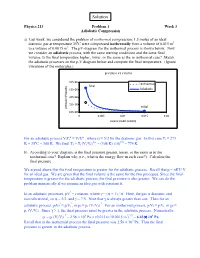
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Thermodynamics of Power Generation
THERMAL MACHINES AND HEAT ENGINES Thermal machines ......................................................................................................................................... 1 The heat engine ......................................................................................................................................... 2 What it is ............................................................................................................................................... 2 What it is for ......................................................................................................................................... 2 Thermal aspects of heat engines ........................................................................................................... 3 Carnot cycle .............................................................................................................................................. 3 Gas power cycles ...................................................................................................................................... 4 Otto cycle .............................................................................................................................................. 5 Diesel cycle ........................................................................................................................................... 8 Brayton cycle ..................................................................................................................................... -

ESCI 241 – Meteorology Lesson 8 - Thermodynamic Diagrams Dr
ESCI 241 – Meteorology Lesson 8 - Thermodynamic Diagrams Dr. DeCaria References: The Use of the Skew T, Log P Diagram in Analysis And Forecasting, AWS/TR-79/006, U.S. Air Force, Revised 1979 An Introduction to Theoretical Meteorology, Hess GENERAL Thermodynamic diagrams are used to display lines representing the major processes that air can undergo (adiabatic, isobaric, isothermal, pseudo- adiabatic). The simplest thermodynamic diagram would be to use pressure as the y-axis and temperature as the x-axis. The ideal thermodynamic diagram has three important properties The area enclosed by a cyclic process on the diagram is proportional to the work done in that process As many of the process lines as possible be straight (or nearly straight) A large angle (90 ideally) between adiabats and isotherms There are several different types of thermodynamic diagrams, all meeting the above criteria to a greater or lesser extent. They are the Stuve diagram, the emagram, the tephigram, and the skew-T/log p diagram The most commonly used diagram in the U.S. is the Skew-T/log p diagram. The Skew-T diagram is the diagram of choice among the National Weather Service and the military. The Stuve diagram is also sometimes used, though area on a Stuve diagram is not proportional to work. SKEW-T/LOG P DIAGRAM Uses natural log of pressure as the vertical coordinate Since pressure decreases exponentially with height, this means that the vertical coordinate roughly represents altitude. Isotherms, instead of being vertical, are slanted upward to the right. Adiabats are lines that are semi-straight, and slope upward to the left. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

Steam Tables and Charts Page 4-1
CHE 2012 Steam Tables and Charts Page 4-1 SECTION 4 STEAM TABLES AND CHARTS WATER AND STEAM Consider the heating of water at constant pressure. If various properties are to be measured, an experiment can be set up where water is heated in a vertical cylinder closed by a piston on which there is a weight. The weight acting down under gravity on a piston of fixed size ensures that the fluid in the cylinder is always subject to the same pressure. Initially the cylinder contains only water at ambient temperature. As this is heated the water changes into steam and certain characteristics may be noted. Initially the water at ambient temperature is subcooled. As heat is added its temperature rises steadily until it reaches the saturation temperature corresponding with the pressure in the cylinder. The volume of the water hardly changes during this process. At this point the water is saturated. As more heat is added, steam is generated and the volume increases dramatically since the steam occupies a greater space than the water from which it was generated. The temperature however remains the same until all the water has been converted into steam. At this point the steam is saturated. As additional heat is added, the temperature of the steam increases but at a faster rate than when the water only was being heated. The Page 4-2 Steam Tables and Charts CHE 2012 volume of the steam also increases. Steam at temperatures above the saturation temperature is superheated. If the temperature T is plotted against the heat added q the three regions namely subcooled water, saturated mixture and superheated steam are clearly indicated. -

Estimation of Internal Pressure of Liquids and Liquid Mixtures
Available online at www.ilcpa.pl International Letters of Chemistry, Physics and Astronomy 5 (2012) 1-7 ISSN 2299-3843 Estimation of internal pressure of liquids and liquid mixtures N. Santhi 1, P. Sabarathinam 2, G. Alamelumangai 1, J. Madhumitha 1, M. Emayavaramban 1 1Department of Chemistry, Government Arts College, C.Mutlur, Chidambaram-608102, India 2Retd. Registrar & Head of the Department of Technology, Annamalai University, Annamalainagar-608002, India E-mail address: [email protected] ABSTRACT By combining the van der Waals’ equation of state and the Free Length Theory of Jacobson, a new theoretical model is developed for the prediction of internal pressure of pure liquids and liquid mixtures. It requires only the molar volume data in addition to the ratio of heat capacities and critical temperature. The proposed model is simple, reliably accurate and capable of predicting internal pressure of pure liquids with an average absolute deviation of 4.24% in the predicted internal pressure values compared to those given in literature. The average absolute deviation in the predicted internal pressure values through the proposed model for the five binary liquid mixtures tested varies from 0.29% to 1.9% when compared to those of literature values. Keywords: van der Waals, pressure of liquids, theory of Jacobson, capacity of heat liquids, equation of Srivastava and Berkowitz 1. INTRODUTION The importance of internal pressure in understanding the properties of liquids and the full potential of internal pressure as a structural probe did become apparent with the pioneering work of Hildebrand 1, 2 and the first review of the subject by Richards 3 appeared in 1925. -

The Carnot Cycle, Reversibility and Entropy
entropy Article The Carnot Cycle, Reversibility and Entropy David Sands Department of Physics and Mathematics, University of Hull, Hull HU6 7RX, UK; [email protected] Abstract: The Carnot cycle and the attendant notions of reversibility and entropy are examined. It is shown how the modern view of these concepts still corresponds to the ideas Clausius laid down in the nineteenth century. As such, they reflect the outmoded idea, current at the time, that heat is motion. It is shown how this view of heat led Clausius to develop the entropy of a body based on the work that could be performed in a reversible process rather than the work that is actually performed in an irreversible process. In consequence, Clausius built into entropy a conflict with energy conservation, which is concerned with actual changes in energy. In this paper, reversibility and irreversibility are investigated by means of a macroscopic formulation of internal mechanisms of damping based on rate equations for the distribution of energy within a gas. It is shown that work processes involving a step change in external pressure, however small, are intrinsically irreversible. However, under idealised conditions of zero damping the gas inside a piston expands and traces out a trajectory through the space of equilibrium states. Therefore, the entropy change due to heat flow from the reservoir matches the entropy change of the equilibrium states. This trajectory can be traced out in reverse as the piston reverses direction, but if the external conditions are adjusted appropriately, the gas can be made to trace out a Carnot cycle in P-V space. -

Analysis of Thermodynamic Processes with Full Consideration of Real Gas Behaviour
Analysis of thermodynamic processes with full consideration of real gas behaviour Version 2.12. - (July 2020) engineering your visions © by B&B-AGEMA, No. 1 Contents • What is TDT ? • How does it look like ? • How does it work ? • Examples engineering your visions © by B&B-AGEMA, No. 2 Introduction What is TDT ? TDT is a Thermodynamic Design Tool, that supports the design and calculation of energetic processes on a 1D thermodynamic approach. The software TDT can be run on Windows operating systems (Windows 7 and higher) or on different LINUX-platforms. engineering your visions © by B&B-AGEMA, No. 3 TDT - Features TDT - Features 1. High calculation accuracy: • real gas properties are considered on thermodynamic calculations • change of state in each component is divided into 100 steps 2. Superior user interface: • ease of input: dialogs on graphic screen • visualized output: graphic system overview, thermodynamic graphs, digital data in tables 3. Applicable various kinds of fluids: • liquid, gas, steam (incl. superheated, super critical point), two-phase state • currently 29 different fluids • user defined fluid mixtures engineering your visions © by B&B-AGEMA, No. 4 How does TDT look like ? engineering your visions © by B&B-AGEMA, No. 5 TDT - User Interface Initial window after program start: start a “New project”, “Open” an existing project, open one of the “Examples”, which are part of the installation, or open the “Manual”. engineering your visions © by B&B-AGEMA, No. 6 TDT - Main Window Structure tree structure action toolbar notebook containing overview and graphs calculation output On the left hand side of the window the entire project information is listed in a tree structure. -

Thermodynamic/Aerological Charts/Diagrams
THERMODYNAMIC/AEROLOGICAL CHARTS/DIAGRAMS 1 /31 • Thermodynamic charts are used to represent the vertical structure of the atmosphere, as well as major thermodynamic processes to which moist air can be subjected. • Thermodynamic charts can be used to obtain easily different thermodynamic properties, e.g. q (potential temperature) and moisture quantities (such as the specific humidity), from a given radiosonde ascent. • Even though today it is possible to compute many quantities directly, thermodynamic diagrams are still very useful and remain videly used. 2 /31 • Each diagram has lines of constant: – p, pressure, – T, temperature, – q, potential temperature, – q, saturation specific humidity. – saturated adiabats. • One difficulty of all diagrams is that they are two dimensional, and the most compact description of the state of the atmosphere encompasses three dimensions, for instance, {T,p,q}. 3 /31 • The simplest and most common form of the aerological diagram has pressure as the ordinate and temperature as the abscissa – the temperature scale is linear – it is usually desirable to have the ordinate approximately representative of height above the surface, thus The ordinate may be proportional to –ln p (the Emagram) or to pR/cp (the Stuve diagram). • The Emagram has the advantage over the Stuve diagram in that area on the diagram is proportional to energy: dw = pdυ = RdT −υdp dp dw = R dT − RT RdT is an exact differential which integrates to zero !∫ !∫ !∫ p !∫ dw = −R!∫ Td(ln p) • A chart with coordinates of T versus ln p has the property of a true thermodynamic diagram, i.e. the area is proportional to energy. -

12/8 and 12/10/2010
PY105 C1 1. Help for Final exam has been posted on WebAssign. 2. The Final exam will be on Wednesday December 15th from 6-8 pm. First Law of Thermodynamics 3. You will take the exam in multiple rooms, divided as follows: SCI 107: Abbasi to Fasullo, as well as Khajah PHO 203: Flynn to Okuda, except for Khajah SCI B58: Ordonez to Zhang 1 2 Heat and Work done by a Gas Thermodynamics Initial: Consider a cylinder of ideal Thermodynamics is the study of systems involving gas at room temperature. Suppose the piston on top of energy in the form of heat and work. the cylinder is free to move vertically without friction. When the cylinder is placed in a container of hot water, heat Equilibrium: is transferred into the cylinder. Where does the heat energy go? Why does the volume increase? 3 4 The First Law of Thermodynamics The First Law of Thermodynamics The First Law is often written as: Some of the heat energy goes into raising the temperature of the gas (which is equivalent to raising the internal energy of the gas). The rest of it does work by raising the piston. ΔEQWint =− Conservation of energy leads to: QEW=Δ + int (the first law of thermodynamics) This form of the First Law says that the change in internal energy of a system is Q is the heat added to a system (or removed if it is negative) equal to the heat supplied to the system Eint is the internal energy of the system (the energy minus the work done by the system (usually associated with the motion of the atoms and/or molecules). -

Pressure Vessels
PRESSURE VESSELS David Roylance Department of Materials Science and Engineering Massachusetts Institute of Technology Cambridge, MA 02139 August 23, 2001 Introduction A good deal of the Mechanics of Materials can be introduced entirely within the confines of uniaxially stressed structural elements, and this was the goal of the previous modules. But of course the real world is three-dimensional, and we need to extend these concepts accordingly. We now take the next step, and consider those structures in which the loading is still simple, but where the stresses and strains now require a second dimension for their description. Both for their value in demonstrating two-dimensional effects and also for their practical use in mechanical design, we turn to a slightly more complicated structural type: the thin-walled pressure vessel. Structures such as pipes or bottles capable of holding internal pressure have been very important in the history of science and technology. Although the ancient Romans had developed municipal engineering to a high order in many ways, the very need for their impressive system of large aqueducts for carrying water was due to their not yet having pipes that could maintain internal pressure. Water can flow uphill when driven by the hydraulic pressure of the reservoir at a higher elevation, but without a pressure-containing pipe an aqueduct must be constructed so the water can run downhill all the way from the reservoir to the destination. Airplane cabins are another familiar example of pressure-containing structures. They illus- trate very dramatically the importance of proper design, since the atmosphere in the cabin has enough energy associated with its relative pressurization compared to the thin air outside that catastrophic crack growth is a real possibility.