Thermodynamic Functions at Isobaric Process of Van Der Waals Gases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
3-1 Adiabatic Compression
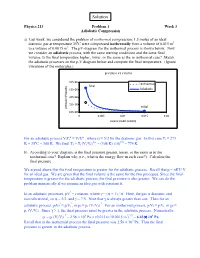
Solution Physics 213 Problem 1 Week 3 Adiabatic Compression a) Last week, we considered the problem of isothermal compression: 1.5 moles of an ideal diatomic gas at temperature 35oC were compressed isothermally from a volume of 0.015 m3 to a volume of 0.0015 m3. The pV-diagram for the isothermal process is shown below. Now we consider an adiabatic process, with the same starting conditions and the same final volume. Is the final temperature higher, lower, or the same as the in isothermal case? Sketch the adiabatic processes on the p-V diagram below and compute the final temperature. (Ignore vibrations of the molecules.) α α For an adiabatic process ViTi = VfTf , where α = 5/2 for the diatomic gas. In this case Ti = 273 o 1/α 2/5 K + 35 C = 308 K. We find Tf = Ti (Vi/Vf) = (308 K) (10) = 774 K. b) According to your diagram, is the final pressure greater, lesser, or the same as in the isothermal case? Explain why (i.e., what is the energy flow in each case?). Calculate the final pressure. We argued above that the final temperature is greater for the adiabatic process. Recall that p = nRT/ V for an ideal gas. We are given that the final volume is the same for the two processes. Since the final temperature is greater for the adiabatic process, the final pressure is also greater. We can do the problem numerically if we assume an idea gas with constant α. γ In an adiabatic processes, pV = constant, where γ = (α + 1) / α. -

Chapter 6 - Engineering Equations of State II
Introductory Chemical Engineering Thermodynamics By J.R. Elliott and C.T. Lira Chapter 6 - Engineering Equations of State II. Generalized Fluid Properties The principle of two-parameter corresponding states 50 50 40 40 30 30 T=705K 20 20 ρ T=286K L 10 10 T=470 T=191K T=329K 0 0 0 0.1 0.2 0.3 0.4 0 0.1 0.2 0.3 0.4 0.5 -10 T=133K -10 ρV Density (g/cc) Density (g/cc) VdW Pressure (bars) in Methane VdW Pressure (bars) in Pentane Critical Definitions: Tc - critical temperature - the temperature above which no liquid can exist. Pc - critical pressure - the pressure above which no vapor can exist. ω - acentric factor - a third parameter which helps to specify the vapor pressure curve which, in turn, affects the rest of the thermodynamic variables. ∂ ∂ 2 P = P = 0 and 0 at Tc and Pc Note: at the critical point, ∂ρ ∂ρ 2 T T Chapter 6 - Engineering Equations of State Slide 1 II. Generalized Fluid Properties The van der Waals (1873) Equation Of State (vdW-EOS) Based on some semi-empirical reasoning about the ways that temperature and density affect the pressure, van der Waals (1873) developed the equation below, which he considered to be fairly crude. We will discuss the reasoning at the end of the chapter, but it is useful to see what the equations are and how we use them before deriving the details. The vdW-EOS is: bρ aρ 1 aρ Z =+1 −= − ()1− bρ RT()1− bρ RT van der Waals’ trick for characterizing the difference between subcritical and supercritical fluids was to recognize that, at the critical point, ∂P ∂ 2 P = 0 and = 0 at T , P ∂ρ ∂ρ 2 c c T T Since there are only two “undetermined parameters” in his EOS (a and b), he has reduced the problem to one of two equations and two unknowns. -

On the Van Der Waals Gas, Contact Geometry and the Toda Chain
entropy Article On the van der Waals Gas, Contact Geometry and the Toda Chain Diego Alarcón, P. Fernández de Córdoba ID , J. M. Isidro * ID and Carlos Orea Instituto Universitario de Matemática Pura y Aplicada, Universidad Politécnica de Valencia, 46022 Valencia, Spain; [email protected] (D.A.); [email protected] (P.F.d.C.); [email protected] (C.O.) * Correspondence: [email protected] Received: 7 June 2018; Accepted: 16 July 2018; Published: 26 July 2018 Abstract: A Toda–chain symmetry is shown to underlie the van der Waals gas and its close cousin, the ideal gas. Links to contact geometry are explored. Keywords: van der Waals gas; contact geometry; Toda chain 1. Introduction The contact geometry of the classical van der Waals gas [1] is described geometrically using a five-dimensional contact manifold M [2] that can be endowed with the local coordinates U (internal energy), S (entropy), V (volume), T (temperature) and p (pressure). This description corresponds to a choice of the fundamental equation, in the energy representation, in which U depends on the two extensive variables S and V. One defines the corresponding momenta T = ¶U/¶S and −p = ¶U/¶V. Then, the standard contact form on M reads [3,4] a = dU + TdS − pdV. (1) One can introduce Poisson brackets on the four-dimensional Poisson manifold P (a submanifold of M) spanned by the coordinates S, V and their conjugate variables T, −p, the nonvanishing brackets being fS, Tg = 1, fV, −pg = 1. (2) Given now an equation of state f (p, T,...) = 0, (3) one can make the replacements T = ¶U/¶S, −p = ¶U/¶V in order to obtain ¶U ¶U f − , ,.. -

Muddiest Point – Entropy and Reversible I Am Confused About Entropy and How It Is Different in a Reversible Versus Irreversible Case
1 Muddiest Point { Entropy and Reversible I am confused about entropy and how it is different in a reversible versus irreversible case. Note: Some of the discussion below follows from the previous muddiest points comment on the general idea of a reversible and an irreversible process. You may wish to have a look at that comment before reading this one. Let's talk about entropy first, and then we will consider how \reversible" gets involved. Generally we divide the universe into two parts, a system (what we are studying) and the surrounding (everything else). In the end the total change in the entropy will be the sum of the change in both, dStotal = dSsystem + dSsurrounding: This total change of entropy has only two possibilities: Either there is no spontaneous change (equilibrium) and dStotal = 0, or there is a spontaneous change because we are not at equilibrium, and dStotal > 0. Of course the entropy change of each piece, system or surroundings, can be positive or negative. However, the second law says the sum must be zero or positive. Let's start by thinking about the entropy change in the system and then we will add the entropy change in the surroundings. Entropy change in the system: When you consider the change in entropy for a process you should first consider whether or not you are looking at an isolated system. Start with an isolated system. An isolated system is not able to exchange energy with anything else (the surroundings) via heat or work. Think of surrounding the system with a perfect, rigid insulating blanket. -

Isothermal Process It Is the Process in Which Other Physical Quantities Might Change but the Temperature of the System Remains Or Is Forced to Remain Constant
Sajit Chandra Shakya Department of Physics Kathmandu Don Bosco College New Baneshwor, Kathmandu Isothermal process It is the process in which other physical quantities might change but the temperature of the system remains or is forced to remain constant. For example, the constant temperature of human body. Under constant temperature, the volume of a gas system is inversely proportional to the pressure applied, the phenomena being called Boyle's Law, written in symbols as 1 V ∝ P 1 Or, V = KB× where KB is a constant quantity. P Or, PV = KB …………….. (i) This means whatever be the values of volume and pressure, their product will be constant. So, P1V1 = KB, P2V2 = KB, P3V3 = KB, etc, Or, P1V1 = P2V2 = P3V3, etc. The requirements for an isothermal process are as follows: 1. The process should be carried very slowly so that there is an ample time for compensation of heat in case of any loss or addition. 2. The boundaries of the system should be highly conducting so that there is a path for heat to flow into or flow away from a closed space in case of any energy loss or oversupply. 3. The boundaries should be made very thin because the resistance of the substance for heat conduction will be less for thin boundaries. Since in an isothermal change, the temperature remains constant, the internal energy also does not change, i.e. dU = 0. So if dQ amount of heat is given to a system which undergoes isothermal change, the relation for the first law of thermodynamics would be dQ = dU + dW Or, dQ = 0 + PdV Or, dQ = PdV This means all the heat supplied will be utilized for performing external work and consequently its value will be very high compared to other processes. -

The First Law of Thermodynamics Continued Pre-Reading: §19.5 Where We Are
Lecture 7 The first law of thermodynamics continued Pre-reading: §19.5 Where we are The pressure p, volume V, and temperature T are related by an equation of state. For an ideal gas, pV = nRT = NkT For an ideal gas, the temperature T is is a direct measure of the average kinetic energy of its 3 3 molecules: KE = nRT = NkT tr 2 2 2 3kT 3RT and vrms = (v )av = = r m r M p Where we are We define the internal energy of a system: UKEPE=+∑∑ interaction Random chaotic between atoms motion & molecules For an ideal gas, f UNkT= 2 i.e. the internal energy depends only on its temperature Where we are By considering adding heat to a fixed volume of an ideal gas, we showed f f Q = Nk∆T = nR∆T 2 2 and so, from the definition of heat capacity Q = nC∆T f we have that C = R for any ideal gas. V 2 Change in internal energy: ∆U = nCV ∆T Heat capacity of an ideal gas Now consider adding heat to an ideal gas at constant pressure. By definition, Q = nCp∆T and W = p∆V = nR∆T So from ∆U = Q W − we get nCV ∆T = nCp∆T nR∆T − or Cp = CV + R It takes greater heat input to raise the temperature of a gas a given amount at constant pressure than constant volume YF §19.4 Ratio of heat capacities Look at the ratio of these heat capacities: we have f C = R V 2 and f + 2 C = C + R = R p V 2 so C p γ = > 1 CV 3 For a monatomic gas, CV = R 3 5 2 so Cp = R + R = R 2 2 C 5 R 5 and γ = p = 2 = =1.67 C 3 R 3 YF §19.4 V 2 Problem An ideal gas is enclosed in a cylinder which has a movable piston. -

Comments on Failures of Van Der Waals' Equation at the Gas–Liquid
Comments on Failures of van der Waals’ Equation at the Gas–Liquid Critical Point, L. V. Woodcock, International Journal of Thermophysics (2018) 39:120 I.H. Umirzakov Institute of Thermophysics, Novosibirsk, Russia [email protected] Abstract These comments are a response to the discussion presented in the above paper concerning the “New comment on Gibbs Density Surface of Fluid Argon: Revised Critical Parameters” by Umirzakov. Here we show that: Woodcock’s results obtained for the dependencies for the isochoric heat capacity, excess Gibbs energy and coexisting difference functional of argon, and coexisting densities of liquid and vapor of the van der Waals fluid and presented in all Figures are incorrect; his Table includes incorrect values of coexisting difference functional; his paper includes many incorrect equations, mathematical and logical errors and physically incorrect assertions concerning the temperature dependences of the isochoric heat capacity and entropy of real fluids; most of the his conclusions are based on the above errors, incorrect data, incorrect comparisons and incorrect dependencies; and most of his conclusions are invalid. We also show that the van der Waals equation of state quantitatively describes the dependencies of saturation pressure on vapor density and temperature near critical point, and the equation of state can describe qualitatively the reduced excess Gibbs energy, rigidity and densities of coexisting liquid and vapor of argon, including the region near critical point. Keywords Coexistence · Critical point · First-order phase transition · Liquid · Phase equilibrium · Vapor 1. Introduction Our comments are a response to a discussion of the article “New comment on Gibbs Density Surface of Fluid Argon: Revised Critical Parameters” by Umirzakov [1] held in the paper [2]. -

Ch 19. the First Law of Thermodynamics
Ch 19. The First Law of Thermodynamics Liu UCD Phy9B 07 1 19-1. Thermodynamic Systems Thermodynamic system: A system that can interact (and exchange energy) with its surroundings Thermodynamic process: A process in which there are changes in the state of a thermodynamic system Heat Q added to the system Q>0 taken away from the system Q<0 (through conduction, convection, radiation) Work done by the system onto its surroundings W>0 done by the surrounding onto the system W<0 Energy change of the system is Q + (-W) or Q-W Gaining energy: +; Losing energy: - Liu UCD Phy9B 07 2 19-2. Work Done During Volume Changes Area: A Pressure: p Force exerted on the piston: F=pA Infinitesimal work done by system dW=Fdx=pAdx=pdV V Work done in a finite volume change W = final pdV ∫V initial Liu UCD Phy9B 07 3 Graphical View of Work Gas expands Gas compresses Constant p dV>0, W>0 dV<0, W<0 W=p(V2-V1) Liu UCD Phy9B 07 4 19-3. Paths Between Thermodynamic States Path: a series of intermediate states between initial state (p1, V1) and a final state (p2, V2) The path between two states is NOT unique. V2 W= p1(V2-V1) +0 W=0+ p2(V2-V1) W = pdV ∫V 1 Work done by the system is path-dependent. Liu UCD Phy9B 07 5 Path Dependence of Heat Transfer Isotherml: Keep temperature const. Insulation + Free expansion (uncontrolled expansion of a gas into vacuum) Heat transfer depends on the initial & final states, also on the path. -

Convolutional Neural Networks for Atomistic Systems
Convolutional neural networks for atomistic systems Kevin Ryczko Department of Physics, University of Ottawa Kyle Mills Department of Physics, University of Ontario Institute of Technology Iryna Luchak Department of Electrical & Computer Engineering, University of British Columbia Christa Homenick National Research Council of Canada Isaac Tamblyn Department of Physics, University of Ontario Institute of Technology Department of Physics, University of Ottawa National Research Council of Canada Abstract We introduce a new method, called CNNAS (convolutional neural networks for atom- istic systems), for calculating the total energy of atomic systems which rivals the com- putational cost of empirical potentials while maintaining the accuracy of ab initio calculations. This method uses deep convolutional neural networks (CNNs), where the input to these networks are simple representations of the atomic structure. We use this approach to predict energies obtained using density functional theory (DFT) for 2D hexagonal lattices of various types. Using a dataset consisting of graphene, hexagonal boron nitride (hBN), and graphene-hBN heterostructures, with and with- out defects, we trained a deep CNN that is capable of predicting DFT energies to an extremely high accuracy, with a mean absolute error (MAE) of 0.198 meV / atom (maximum absolute error of 16.1 meV / atom). To explore our new methodology, arXiv:1706.09496v2 [cond-mat.mtrl-sci] 20 Mar 2018 Email addresses: [email protected] (Kevin Ryczko), [email protected] (Isaac Tamblyn) Preprint submitted to Computational Materials Science March 21, 2018 we investigate the ability of a deep neural network (DNN) in predicting a Lennard- Jones energy and separation distance for a dataset of dimer molecules in both two and three dimensions. -

Thermodynamic Propertiesproperties
ThermodynamicThermodynamic PropertiesProperties PropertyProperty TableTable -- from direct measurement EquationEquation ofof StateState -- any equation that relates P,v, and T of a substance jump ExerciseExercise 33--1212 A bucket containing 2 liters of R-12 is left outside in the atmosphere (0.1 MPa) a) What is the R-12 temperature assuming it is in the saturated state. b) the surrounding transfer heat at the rate of 1KW to the liquid. How long will take for all R-12 vaporize? See R-12 (diclorindifluormethane) on Table A-2 SolutionSolution -- pagepage 11 Part a) From table A-2, at the saturation pressure of 0.1 MPa one finds: • Tsaturation = - 30oC 3 • vliq = 0.000672 m /kg 3 • vvap = 0.159375 m /kg • hlv = 165KJ/kg (vaporization heat) SolutionSolution -- pagepage 22 Part b) The mass of R-12 is m = Volume/vL, m = 0.002/0.000672 = 2.98 kg The vaporization energy: Evap = vap energy * mass = 165*2.98 = 492 KJ Time = Heat/Power = 492 sec or 8.2 min GASGAS PROPERTIESPROPERTIES Ideal -Gas Equation of State M PV = nR T; n = u mol Universal gas constant is given on Ru = 8.31434 kJ/kmol-K = 8.31434 kPa-m3/kmol-k = 0.0831434 bar-m3/kmol-K = 82.05 L-atm/kmol-K = 1.9858 Btu/lbmol-R = 1545.35 ft-lbf/lbmol-R = 10.73 psia-ft3/lbmol-R EExamplexample Determine the particular gas constant for air (28.97 kg/kmol) and hydrogen (2.016 kg/kmol). kJ 8.1417 kJ = Ru = kmol − K = 0.287 Rair kg M 28.97 kg − K kmol kJ 8.1417 kJ = kmol − K = 4.124 Rhydrogen kg 2.016 kg − K kmol IdealIdeal GasGas “Law”“Law” isis aa simplesimple EquationEquation ofof StateState PV = MRT Pv = RT PV = NRuT P V P V 1 1 = 2 2 T1 T2 QuestionQuestion …...….. -

Chemical Engineering Thermodynamics
CHEMICAL ENGINEERING THERMODYNAMICS Andrew S. Rosen SYMBOL DICTIONARY | 1 TABLE OF CONTENTS Symbol Dictionary ........................................................................................................................ 3 1. Measured Thermodynamic Properties and Other Basic Concepts .................................. 5 1.1 Preliminary Concepts – The Language of Thermodynamics ........................................................ 5 1.2 Measured Thermodynamic Properties .......................................................................................... 5 1.2.1 Volume .................................................................................................................................................... 5 1.2.2 Temperature ............................................................................................................................................. 5 1.2.3 Pressure .................................................................................................................................................... 6 1.3 Equilibrium ................................................................................................................................... 7 1.3.1 Fundamental Definitions .......................................................................................................................... 7 1.3.2 Independent and Dependent Thermodynamic Properties ........................................................................ 7 1.3.3 Phases ..................................................................................................................................................... -

12/8 and 12/10/2010
PY105 C1 1. Help for Final exam has been posted on WebAssign. 2. The Final exam will be on Wednesday December 15th from 6-8 pm. First Law of Thermodynamics 3. You will take the exam in multiple rooms, divided as follows: SCI 107: Abbasi to Fasullo, as well as Khajah PHO 203: Flynn to Okuda, except for Khajah SCI B58: Ordonez to Zhang 1 2 Heat and Work done by a Gas Thermodynamics Initial: Consider a cylinder of ideal Thermodynamics is the study of systems involving gas at room temperature. Suppose the piston on top of energy in the form of heat and work. the cylinder is free to move vertically without friction. When the cylinder is placed in a container of hot water, heat Equilibrium: is transferred into the cylinder. Where does the heat energy go? Why does the volume increase? 3 4 The First Law of Thermodynamics The First Law of Thermodynamics The First Law is often written as: Some of the heat energy goes into raising the temperature of the gas (which is equivalent to raising the internal energy of the gas). The rest of it does work by raising the piston. ΔEQWint =− Conservation of energy leads to: QEW=Δ + int (the first law of thermodynamics) This form of the First Law says that the change in internal energy of a system is Q is the heat added to a system (or removed if it is negative) equal to the heat supplied to the system Eint is the internal energy of the system (the energy minus the work done by the system (usually associated with the motion of the atoms and/or molecules).