Surface Chemistry and Spectroscopy of Chromium in Inorganic Oxides
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
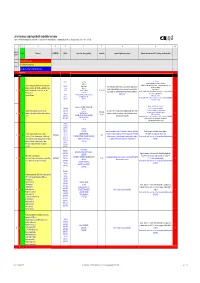
List of Substances Subject to Prohibited / Undesirable / Declaration
List of substances subject to prohibited / undesirable / declaration Appendix 1 to the Guidelines to Design and Purchasing Prohibition of Harmful Substances : ebmpapstMulfingen: 90-001 ebmpapstSt.Georgen: WN 3-12.2 ebmpapstLandshut: 60118.00040 (Ver. 5.0-31.07.2008) 1 2 3 4 5 6 7 8 9 10 Consecu N - new Substance EU-INDEX-No.: CAS-No. Source (law, decree, guideline). Hazard/risk example of application/occurrence Remarks and comments (of 0.1% deviating consideration limit) tive No. V = prohibited (verboten) U = undesirable (unerwünscht) D = subject to declaration (deklarationspflichtig) V = prohibited RoHS from 100 ppm 7439-92-1 TRGS 905, Pb-stabilisers and pigments are subject to declaration. GefStoffV, Prohibited: Anhydrite neutral lead carbonate, lead hydrogen carbonate and lead Lead and compounds (red lead oxide, lead sulphate, lead (7446-14-2 ChemVerbotsV, cable coating, heat transfer medium for accumulators, hybrid integrated sulphate as dye pigment hydrogen carbonate, lead carbonate, lead phtalates, lead 1319-46-6 BatterieV, circuits, stabilisers in plastics, vessels and pipes for aggressive liquids, < 0.4 % in batteries 1 V N acetates, lead phosphates, lead stereates , etc) lead 598-63-0 2002/95/EG (RoHS), K3, R 1, R 3 V E F according to RoHS: Lead as alloying additive in aluminum (up to 0.4 mass%) in steel (up to 67/458/EEC pigment production, lead alloys, anti-corrosion agents (fuel additives), chromate: refer to 0.35 mass%), in copper (up to 4%), 301-04-2 76/769/EEC (89/667/EEC, 97/10/EC, 97/56/EC) soldering agent chromium (VI) -

Sd0000029 Evaluation of Chromfte Ore and The
SD0000029 EVALUATION OF CHROMFTE ORE AND THE OPTIMUM METHODS FOR INDUSTRIAL EXTRACTION OF CHROMIUM A TI1HSIS SUBMITTED BY Bakheit Mustafa Mohamed Salih IN CANDIDATURE FOR TIU:DI:GRFJ:OI MASTER OF SC1HNCT DEPARTMENT OF CHEMISTRY FACULTY OF SCIENCE (P.O.BOX 321) OCTOBER 1999 UNIVERSITY OF KHARTOUM 31/ 28 ABSTRACT Samples of chromite ore, collected from Gam and Cheikay mining area (Ingessana Hills) in east Sudan, were analysed to assess the chromium content. Methods for extraction and analysis of chromium metal were developed and established. Analysis were carried out using atomic absorption spectroscopy (AAS) to estimate the contents of chromium, iron, calcium, and magnesium. X-ray fluorescence (XRF) was used to evaluate the levels of chromium, iron, and calcium in the ore. Volumetric analysis was performed to assess chromium and iron, whilst gravimetric analysis was employed to measure the amounts of calcium, magnesium, aluminum and silicon present in the ore. The data was chemically and statistically analyzed to compare the results obtained by the given analytical methods. The results are in good agreement except iron oxide, which displayed a significantly different value when measured by x-ray fluorescence. The data obtained exhibited similarity in almost all cases, when compared with local and global researches, reports, and literature. The study has revealed the average contents of Cr2O3, FeO, CaO, MgO, A12O3, and SiO2 as 40.66. 11.96, 11.94. 0.36. 16.94, 11.45% respectively. MnO and NiO were detected in trace amounts, the corresponding levels in the ore being 72 and 27 ppm. The average chromium content in extracted potassium dichromate measured by using AAS, XRF, and volumetric methods was found to be 31.7%. -

Potassium Dichromate CAS Number: 7778-50-9 EC Number: 231-906-6
21.5.2010 COMMENTS AND RESPONSE TO COMMENTS ON ANNEX XV SVHC: PROPOSAL AND JUSTIFICATION Substance name: Potassium dichromate CAS number: 7778-50-9 EC number: 231-906-6 Reason of the submission of the Annex XV: CMR Disclaimer: The European Chemicals Agency is not responsible for the content of this document. The Response to Comments table has been prepared by the competent authority of the Member State preparing the proposal for identification of a Substance of Very High Concern. The comments were received during the public consultation of the Annex XV dossier. General comments Date Submitted by (name, Comment Response Organisation/MSCA) 20100323 CLEAPSS, UK (on behalf of an CLEAPSS is an advisory service providing support in No answer expected. organisation) science and technology for a consortium of local authorities and their schools including establishments for pupils with special needs. Independent schools, post-16 colleges, teacher training establishments, curriculum developers and others can apply for associate membership. We offer help from nursery education through to A-level studies or equivalent. Our services cover health & safety, risk assessment, sources and use of chemicals, living organisms and equipment. CLEAPSS also provides advice on technicians & their jobs, as well as the design of laboratories and facilities & fittings for D&T and science rooms. 20100324 Germany, company (on behalf of an We reject the ban of Potassium Dichromate in general, Thank for your comment. organisation) because the substance is used in a low concentration (< 1%) in an almost closed system of cuvette tests 20100408 Draeger Safety AG & Co. KGaA, comments k2Cr2O7R1.doc Thank you for this information. -

Rpt POL-TOXIC AIR POLLUTANTS 98 BY
SWCAA TOXIC AIR POLLUTANTS '98 by CAS ASIL TAP SQER CAS No HAP POLLUTANT NAME HAP CAT 24hr ug/m3 Ann ug/m3 Class lbs/yr lbs/hr none17 BN 1750 0.20 ALUMINUM compounds none0.00023 AY None None ARSENIC compounds (E649418) ARSENIC COMPOUNDS none0.12 AY 20 None BENZENE, TOLUENE, ETHYLBENZENE, XYLENES BENZENE none0.12 AY 20 None BTEX BENZENE none0.000083 AY None None CHROMIUM (VI) compounds CHROMIUM COMPOUN none0.000083 AY None None CHROMIUM compounds (E649962) CHROMIUM COMPOUN none0.0016 AY 0.5 None COKE OVEN COMPOUNDS (E649830) - CAA 112B COKE OVEN EMISSIONS none3.3 BN 175 0.02 COPPER compounds none0.67 BN 175 0.02 COTTON DUST (raw) none17 BY 1,750 0.20 CYANIDE compounds CYANIDE COMPOUNDS none33 BN 5,250 0.60 FIBROUS GLASS DUST none33 BY 5,250 0.60 FINE MINERAL FIBERS FINE MINERAL FIBERS none8.3 BN 175 0.20 FLUORIDES, as F, containing fluoride, NOS none0.00000003 AY None None FURANS, NITRO- DIOXINS/FURANS none5900 BY 43,748 5.0 HEXANE, other isomers none3.3 BN 175 0.02 IRON SALTS, soluble as Fe none00 AN None None ISOPROPYL OILS none0.5 AY None None LEAD compounds (E650002) LEAD COMPOUNDS none0.4 BY 175 0.02 MANGANESE compounds (E650010) MANGANESE COMPOU none0.33 BY 175 0.02 MERCURY compounds (E650028) MERCURY COMPOUND none33 BY 5,250 0.60 MINERAL FIBERS ((fine), incl glass, glass wool, rock wool, slag w FINE MINERAL FIBERS none0.0021 AY 0.5 None NICKEL 59 (NY059280) NICKEL COMPOUNDS none0.0021 AY 0.5 None NICKEL compounds (E650036) NICKEL COMPOUNDS none0.00000003 AY None None NITROFURANS (nitrofurans furazolidone) DIOXINS/FURANS none0.0013 -

Chromate and the Environment: Removal and Utilization of Industrial Waste
J. Chem. Chem. Eng. 10 (2016) 147-152 doi: 10.17265/1934-7375/2016.03.006 D DAVID PUBLISHING Chromate and the Environment: Removal and Utilization of Industrial Waste Fernando B. Mainier, Pedro Paulo B. Leite, Marcone F. Reis and Thiago Teobaldo Silva Engineering School, Federal Fluminense University(UFF), Niterói, Rio de Janeiro 24220-261, Brazil Abstract: Chromate and dichromate sodium as a function of oxidizer characteristics are used in several industrial areas; for example, in surface protection of coated parts of cadmium, zinc and aluminum (chromate coated treated), corrosion inhibitors, the treatment of leather, the manufacture of pigments, etc. However, the use of such products has been questioned due to the problems of toxicity and pollution that can be caused in the environmental. The Brazilian environmental agency has established that the concentrations of 2- chromate in water courses are less than 0.5 ppm. In order to reuse chromate (CrO4 ) from industrial effluent, laboratory experiments have been proposed based on chemical reduction or electrolytic processes, in order to transform these chromate ions in a final mix of oxides (in solid form), which can then be packed and sent to the production process of sodium chromate. The results of these experiments have become useful industrially (without regard to costs) considering the environmental reuse and the life cycle of the chemical compound. Key words: Chromate, dichromate, contamination, chemical reduction, electrolytic process. 1. Introduction waste, among others [1, 2]. In -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

CHROMIUM and Ehromium Eompounds
CHROMIUM AND eHROMIUM eOMPOUNDS Chromium and chromium compounds were considered by previous IAC Working Groups, in 1972, 1979, 1982 and 1987 (lARe, 1973, 1979, 1980a, 1982, 1987a). Since that time, new data have become available, and these are included in the present monograph and have been taken into consideration in the evaluation. 1. ehemical and Physical Data The list of chromium alloys and compounds given in Table 1 is not exhaustive, nor does it necessarily reflect the commercial importance of the various chro- mium-containing substances, but it is indicative of the range of chromium alloys and compounds available. 1.1 Synonyms, trade names and molecular formulae of chromium and selected chromium-containing compouDds Table 1. Synonyms (Chemical Abstracts Service names are given iD bold), trade names and atomic or molecular formulae of chromium and selected chromium compounds Chemical Chem. Abstr. Synonyms and trade names Formulab na me Seivices Reg. No. a Metallc chromium (0) and chromium (0) alloys Chromium 7440-47-3 Chrome Cr Cobalt-chro- 11114-92-4 Chromium alloy (nonbase), Co, Cr; cobalt alloy (non- mium alloyC (91700-55-9) base), Co, Cr Cobalt-chro- 12629-02.; Cobalt alloy (base), Co 56-68, Cr 25-29, Mo 5-6, Ni mium-molyb- (8064-15-1; 1.8-3.8, Fe 0-3, Mn 0-1, Si 0-1, C 0.2-0.3 (ASTM A567-1) denum 11068-92-1; alloyC 12618-69-8; 55345-18- 1; -49- 50 lARe MONOGRAHS VOLUME 49 Table 1 (contd) Chernical Chern. Abstr. Synonyrs and trade narnes Forrulab narne Servces Reg. -

Colorimetric Studies on the Nature of Chromate Solutions
COLORIMETRIC STUDIES ON NATURE OF CHROMATE SOLUTIONS. 829 [CONTRIBUTIONSFROM THE CHEMICAL LABORATORY OF THE UNIVERSITY OF WASH- INGTON. ] COLORIMETRIC STUDIES ON THE NATURE OF CHROMATE SOLUTIONS. BY WILLIAMM. DEAN Received January 17. 1914 During the past quarter of a century many investigations have been made to determine the constitution and equilibria of chromates in aqueous solution. The preponderance of evidence previously and now contributed strengthens the conclusion that the most important equilibrium may be expressed by the equation : H2Cr207+ H20 zHzCr04. As will be shown in the following experiments, the colors’ of chromic acid, dichromates and chromates in solutions more dilute than 0.0037~are identical, that is, at these concentrations, both the chromic acid and the dichromates are completely2 hydrolyzed, in accordance with the above equation. Furthermore, it will be shown that heat3 promotes the reversal of such re- actions, therefore, since the effect of hydroxyl and hydrogen ions has long been known, we have the following simple view of chromate solutions : Concentration Heat (CrO& I_ HzCrrOla Acids 1 Dilution H2Cr3Olo HtCrzO, HZCr04 1 Cold ( Alkalies These reactions are assumed to involve only hydrolytic equilibria; however, when chromic oxide is dissolved in water and alkalies in varied proportions, many equilibria may be involved as typified by: hydrolysis Jr neutralization hydration dehydration (condensation) ionization 1_ association Neutralization. When chromic oxide is partially or fully neutralized with alkali, or is treated with an excess of the same (concentration and temperature re- maining unchanged) the following reactions4 are possible : (a) 12CrOs f 3KOH gKHCrdOls 4(b) 12CrOa + 6KOH 3KzCr401s 4- 3Ht0 Ostwald (2. -

Synthesis and Characterization of Novel Nanofiber Based Calixarene
J Incl Phenom Macrocycl Chem (2016) 85:49–58 DOI 10.1007/s10847-016-0604-5 ORIGINAL ARTICLE Synthesis and characterization of novel nanofiber based calixarene and its binding efficiency towards chromium and uranium ions 1,2 3 1 Fatih O¨ zcan • Mevlu¨t Bayrakcı • S¸ eref Ertul Received: 19 January 2016 / Accepted: 11 March 2016 / Published online: 18 March 2016 Ó Springer Science+Business Media Dordrecht 2016 Abstract The objective of this study is to obtain Introduction nanofibrous precursor based calixarene with high ion adsorption capacity by electrospinning of blended solution Nanotechnology has gained interest in recent years from of polyacrylonitrile (PAN) and upper rim functionalized both public and private sector organizations. Therefore, calix[4]arene bearing N-methylglucamine (Calix-NMG). nanotechnology has offered several novel products, which The obtained electrospun fibers were characterized using have superior properties making them valuable for a wide fourier transform infrared (FT-IR-ATR), scanning electron range of applications. One of the major successes of nan- microscope (SEM) equipped with energy-dispersive X-ray otechnology has been nanofibers. Nanofibers can be man- spectrometry (EDX) and thermogravimetric analyses (TGA ufactured by electrospinning which is a technique to obtain and DSC). Analysis indicated that preparation of nanofi- fiber structures that can range in diameter from nanometers bers based Calix-NMG was successfully achieved. The ion to micrometers [1–4]. Electrospun fibers can be made of binding studies exhibited that the nanofiber based Calix- many types of polymers and composite combinations that NMG could be efficiently used for the binding of chromate can be utilized to imitate properties of native extracellular anions and uranium cations. -

I26 X-ESR-G-00048 Rev 2
X-ESR-G-00048 Revision 2 Page 1 of 26 Keywords: Mercury, Monomethyl, Dimethyl, Methylation, Speciation, Solubility, Flammability. Volatilization and Flammability Characteristics of Elemental and Organic Mercury Heather J. Meraw June 2015 Savannah River Site Aiken, SC 29808 www.srremediation.com X-ESR-G-00048 Revision 2 Page 2 of 26 Table of Contents 1 Summary of Revisions .............................................................................................................................. 4 2 Purpose ....................................................................................................................................................... 4 3 Introduction ............................................................................................................................................... 7 4 Executive Summary .................................................................................................................................. 7 5 Assumptions .............................................................................................................................................. 7 6 Discussion .................................................................................................................................................. 9 6.1 Mercury Compounds ........................................................................................................................ 9 6.2 Volatility ........................................................................................................................................... -

NBO Applications, 2008
NBO 2008 (Jan-Dec) - 910 references Compiled by Emily Wixson; Updated by Ariel Neff 4/16/13 Adalsteinsson, H.; Debusschere, B. J.; Long, K. R.; Najm, H. N. Components for atomistic-to-continuum multiscale modeling of flow in micro- and nanofluidic systems Scientific Programming, (16): 297-313 2008. Adcock, W.; Trout, N. A. Diastereofacial selectivity in some 4-substituted (X) 2-adamantyl derivatives: electronic versus steric effects Journal of Physical Organic Chemistry, (21): 68-72 2008. Agapito, F.; Nunes, P. A.; Costa Cabral, B. J.; Borges dos Santos, R. A.; Martinho Simoes, J. A. Energetic differences between the five- and six-membered ring hydrocarbons: Strain energies in the parent and radical molecules Journal of Organic Chemistry, (73): 6213-6223 2008. Aguilar-Castro, L.; Tlahuextl, M.; Mendoza-Huizar, L. H.; Tapia-Benavides, A. R.; Tlahuext, H. Hydrogen bond studies in substituted N-(2-hydroxyphenyl)-2-[(4- methylbenzenesulfonyl)amino]acetamides Arkivoc: 210-226 2008. Alajarin, M.; Cabrera, J.; Pastor, A.; Sanchez-Andrada, P.; Bautista, D. Polar hetero-Diels-alder reactions of 4-alkenylthiazoles with 1,2,4-triazoline-3,5-diones: An experimental and computational study Journal of Organic Chemistry, (73): 963-973 2008. Albertin, G.; Antoniutti, S.; Baldan, D.; Castro, J.; Garcia-Fontan, S. Preparation of benzyl azide complexes of iridium(III) Inorganic Chemistry, (47): 742-748 2008. Alcoba, D. R.; Ona, O. Determination of energies and electronic densities of functional groups according to partitionings in the physical space Journal of Physical Chemistry A, (112): 10023-10028 2008. Alia, J. D.; Vlaisavljevich, B. Prediction of molecular properties including symmetry from quantum-based molecular structural formulas, VIF Journal of Physical Chemistry A, (112): 9784-9795 2008. -

TR-546: Sodium Dichromate Dihydrate (CASRN 7789-12-0)
NTP TECHNICAL REPORT ON THE TOXICOLOGY AND CARCINOGENESIS STUDIES OF SODIUM DICHROMATE DIHYDRATE (CAS NO. 7789-12-0) IN F344/N RATS AND B6C3F1 MICE (DRINKING WATER STUDIES) NATIONAL TOXICOLOGY PROGRAM P.O. Box 12233 Research Triangle Park, NC 27709 July 2008 NTP TR 546 NIH Publication No. 08-5887 National Institutes of Health Public Health Service U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES FOREWORD The National Toxicology Program (NTP) is an interagency program within the Public Health Service (PHS) of the Department of Health and Human Services (HHS) and is headquartered at the National Institute of Environmental Health Sciences of the National Institutes of Health (NIEHS/NIH). Three agencies contribute resources to the program: NIEHS/NIH, the National Institute for Occupational Safety and Health of the Centers for Disease Control and Prevention (NIOSH/CDC), and the National Center for Toxicological Research of the Food and Drug Administration (NCTR/FDA). Established in 1978, the NTP is charged with coordinating toxicological testing activities, strengthening the science base in toxicology, developing and validating improved testing methods, and providing information about potentially toxic substances to health regulatory and research agencies, scientific and medical communities, and the public. The Technical Report series began in 1976 with carcinogenesis studies conducted by the National Cancer Institute. In 1981, this bioassay program was transferred to the NTP. The studies described in the Technical Report series are designed and conducted to characterize and evaluate the toxicologic potential, including carcinogenic activity, of selected substances in laboratory animals (usually two species, rats and mice). Substances selected for NTP toxicity and carcinogenicity studies are chosen primarily on the basis of human exposure, level of production, and chemical structure.