Executive Summary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Hexavalent Chromium Hexavalent
Toxic Heavy Metals Series Toxic Heavy Metals Hexavalent ChromiumHexavalent This document will familiarize the reader with the toxic metal Hexavalent Chromium, OSHA standards, and methods for prevention and decontamination. Hygenall Corporation 6275 University Drive, STE 37-390 Huntsville, AL 35806 256.781.4486 [email protected] Toxic Metal Series Hexavalent Chromium Contents Introduction . 3 Worker Exposure and Health Consequences . 4 OSHA’s Hexavalent Chromium Standards . 4 Exposure Limits . 4 Exposure Monitoring and Determinations . 4 Scheduled Monitoring Option . 5 Regulated Areas . 6 Control Measures . 7 Requirements for Protective Work Clothing and Equipment . 8 Hygiene Areas and Practices . 9 Housekeeping . 10 Recommended Cleaning and Decontamination Products…11 Medical Surveillance . 12 Worker Training and Communication . 13 Recordkeeping . 14 Effective Dates . 14 Additional Information . 15 References . 16 2 | P a g e Hexavalent Chromium Toxic Metal Series INTRODUCTION This document is intended to give readers an overview of the provisions and requirements of the OSHA Hexavalent Chromium standards for general industry (29 CFR 1910.1026), shipyards (29 CFR 1915.1026), and construction (29 CFR 1926.1126). Hexavalent chromium (Cr(VI)) is a toxic form of the element chromium. Hexavalent chromium is rarely found in nature and is generally man-made. Cr(VI) is widely used in pigments, metal finishing electroplating), wood preservatives and fungicides, and in chemical synthesis as an ingredient and catalyst. Table 1 lists some selected Cr(VI) compounds with their synonyms and common uses. Hexavalent chromium may also be present in fumes generated during the production or welding of chrome alloys. Chromium metal is often alloyed with other metals or plated on metal and plastic substrates to improve corrosion resistance and provide protective coatings. -

Chromium Compounds Hazard Summary
Chromium Compounds Hazard Summary Chromium occurs in the environment primarily in two valence states, trivalent chromium (Cr III) and hexavalent chromium (Cr VI). Exposure may occur from natural or industrial sources of chromium. Chromium III is much less toxic than chromium (VI). The respiratory tract is also the major target organ for chromium (III) toxicity, similar to chromium (VI). Chromium (III) is an essential element in humans. The body can detoxify some amount of chromium (VI) to chromium (III). The respiratory tract is the major target organ for chromium (VI) toxicity, for acute (short-term) and chronic (long-term) inhalation exposures. Shortness of breath, coughing, and wheezing were reported from a case of acute exposure to chromium (VI), while perforations and ulcerations of the septum, bronchitis, decreased pulmonary function, pneumonia, and other respiratory effects have been noted from chronic exposure. Human studies have clearly established that inhaled chromium (VI) is a human carcinogen, resulting in an increased risk of lung cancer. Animal studies have shown chromium (VI) to cause lung tumors via inhalation exposure. Please Note: The main sources of information for this fact sheet are EPA's Integrated Risk Information System (IRIS) (7), which contains information on inhalation chronic toxicity and the RfC and oral chronic toxicity and the RfD, and the carcinogenic effects of chromium including the unit cancer risk for inhalation exposure, EPA's Toxicological Review of Trivalent Chromium and Toxicological Review of Hexavalent Chromium (3), and the Agency for Toxic Substances and Disease Registry's (ATSDR's) Toxicological Profile for Chromium. (1) Uses The metal chromium is used mainly for making steel and other alloys. -

Beneficial Reuse and Waste Minimization of Hexavalent Chrome
Hexavalent Chromium Treatment Technologies Research and Development Office Science and Technology Program Final Report ST-2018-9085-01 U.S. Department of the Interior Bureau of Reclamation Research and Development Office 10/1/2018 Mission Statements Protecting America's Great Outdoors and Powering Our Future The Department of the Interior protects and manages the Nation's natural resources and cultural heritage; provides scientific and other information about those resources; and honors its trust responsibilities or special commitments to American Indians, Alaska Natives, and affiliated island communities. The following form is a Standard form 298, Report Documentation Page. This report was sponsored by the Bureau of Reclamations Research and Development office. For more detailed information about this Report documentation page please contact Miguel Arias-Paic at 303-445-2131. THIS TEXT WILL BE INVISIBLE. IT IS FOR 508 COMPLIANCE OF THE NEXT PAGE. Disclaimer Information in this report may not be used for advertising or promotional purposes. The data and findings should not be construed as an endorsement of any product or firm by the Bureau of Reclamation, Department of Interior, or Federal Government. The products evaluated in the report were evaluated for purposes specific to the Bureau of Reclamation mission. Reclamation gives no warranties or guarantees, expressed or implied, for the products evaluated in this report, including merchantability or fitness for a particular purpose. Form Approved REPORT DOCUMENTATION PAGE OMB No. 0704-0188 T1. REPORT DATE: T2. REPORT TYPE: T3. DATES COVERED SEPTEMBER 2018 RESEARCH T4. TITLE AND SUBTITLE 5a. CONTRACT NUMBER Hexavalent Chromium Treatment Technologies Z1759, X9085, FA280 5b. -

Jason Lotter, MS, CIH
Jason Lotter, MS, CIH Current Position Summary of Experience Supervising Health Mr. Jason Lotter is a Supervising Health Scientist with Cardno ChemRisk in Chicago. His Scientist principal areas of training and expertise include industrial hygiene, exposure and risk assessment, and occupational safety. He has been involved in researching, measuring, Discipline Areas and reconstructing exposures, and assessing risks to consumers, residents, and workers, > Industrial Hygiene from a variety of chemicals including formaldehyde, silica, diacetyl, coal dust, lead, > Exposure particulate matter, hexavalent chromium, asbestos, and metals. Mr. Lotter completed his Assessment master’s degree in Public Health Science, with an emphasis in industrial hygiene, from > Risk Assessment the University of Illinois at Chicago. For his master’s thesis, he evaluated groundwater > Occupational Safety arsenic in Guatemala and conducted a risk assessment related to arsenic exposure for local residents. Years' Experience 7 Years At ChemRisk, Mr. Lotter has conducted independent research on various topics and chemicals, and has presented his research at scientific conferences and published in the Joined Cardno peer-reviewed literature. He has also conducted indoor air modeling for formaldehyde, 2013 provided industrial hygiene and occupational safety support during refinery turnarounds, and has assisted in the completion of various exposure simulation studies. Education > MS, Public Health Mr. Lotter currently co-leads the Cardno ChemRisk Cleaning and Disinfectant Product Science, University initiative, focusing on peracetic acid and other antimicrobial chemicals. He is a member of of Illinois at Chicago, the American Industrial Hygiene Association (AIHA) and currently serves on the Board of 2012 Directors of the Chicago Local Section of AIHA. He has been as a Certified Industrial > BS, Civil Hygienist (CIH) since 2016. -

Hexavalent and Total Chromium at Low Reporting Concentrations in Source-Water Aquifers and Surface Waters Used for Public Supply in Illinois, 2013
Prepared in cooperation with the Illinois Environmental Protection Agency Hexavalent and Total Chromium at Low Reporting Concentrations in Source-Water Aquifers and Surface Waters Used for Public Supply in Illinois, 2013 Scientific Investigations Report 2015–5020 U.S. Department of the Interior U.S. Geological Survey Front cover and other images: Front cover shows collection of groundwater samples, Union-York Water District (intake 00251). This and other photography by Patrick Mills, U.S. Geological Survey. Hexavalent and Total Chromium at Low Reporting Concentrations in Source-Water Aquifers and Surface Waters Used for Public Supply in Illinois, 2013 By Patrick C. Mills and Richard P. Cobb Prepared in cooperation with the Illinois Environmental Protection Agency Scientific Investigations Report 2015–5020 U.S. Department of the Interior U.S. Geological Survey U.S. Department of the Interior SALLY JEWELL, Secretary U.S. Geological Survey Suzette M. Kimball, Acting Director U.S. Geological Survey, Reston, Virginia: 2015 For more information on the USGS—the Federal source for science about the Earth, its natural and living resources, natural hazards, and the environment—visit http://www.usgs.gov or call 1–888–ASK–USGS. For an overview of USGS information products, including maps, imagery, and publications, visit http://www.usgs.gov/pubprod/. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. Although this information product, for the most part, is in the public domain, it also may contain copyrighted materials as noted in the text. Permission to reproduce copyrighted items must be secured from the copyright owner. -

Low-Level Hexavalent Chromium Treatment Options: Bench-Scale Evaluation
Tailored Collaboration Low-Level Hexavalent Chromium Treatment Options: Bench-Scale Evaluation Subject Area: High-Quality Water Low-Level Hexavalent Chromium Treatment Options: Bench-Scale Evaluation ©2004 AwwaRF. All rights reserved. About the Awwa Research Foundation The Awwa Research Foundation (AwwaRF) is a member-supported, international, nonprofit organization that sponsors research to enable water utilities, public health agencies, and other professionals to provide safe and affordable drinking water to consumers. The Foundation's mission is to advance the science of water to improve the quality of life. To achieve this mission, the Foundation sponsors studies on all aspects of drinking water, including supply and resources, treatment, monitoring and analysis, distribution, management, and health effects. Funding for research is provided primarily by subscription payments from approximately 1,000 utilities, consulting firms, and manufacturers in North America and abroad. Additional funding comes from collaborative partnerships with other national and international organizations, allowing for resources to be leveraged, expertise to be shared, and broad-based knowledge to be developed and disseminated. Government funding serves as a third source of research dollars. From its headquarters in Denver, Colorado, the Foundation's staff directs and supports the efforts of more than 800 volunteers who serve on the board of trustees and various committees. These volunteers represent many facets of the water industry, and contribute their expertise to select and monitor research studies that benefit the entire drinking water community. The results of research are disseminated through a number of channels, including reports, the Web site, conferences, and periodicals. For subscribers, the Foundation serves as a cooperative program in which water suppliers unite to pool their resources. -

Waste Discharge Requirements for Pacific Gas and Electric Company Groundwater Remediation Project
CALIFORNIA REGIONAL WATER QUALITY CONTROL BOARD LAHONTAN REGION BOARD ORDER NO. R6V-2014-0023 WASTE DISCHARGE REQUIREMENTS FOR PACIFIC GAS AND ELECTRIC COMPANY GROUNDWATER REMEDIATION PROJECT AGRICULTURAL TREATMENT UNITS WDID NO. 6B361403002 _________________________San Bernardino County______________________ The California Regional Water Quality Control Board, Lahontan Region (Water Board), finds: 1. Discharger Pacific Gas and Electric Company (PG&E) is the owner and operator of the natural gas compressor station in Hinkley where hexavalent chromium was discharged from historical waste water releases to groundwater. For the purposes of this Order, PG&E is referred to as the “Discharger.” This Water Board Order (Order) supersedes and rescinds the previous Order No. R6V-2004-0034 and amendments, and Investigative Order R6V-2011-0078. 2. Groundwater Contamination The compressor station began operating in 1952 and discharged untreated cooling tower water containing hexavalent chromium to unlined ponds until 1964. Wastewater then percolated through soil to the water table, approximately 80 feet below, creating a chromium plume in groundwater. Since 1991, PG&E has implemented various interim remediation projects to clean up chromium in groundwater at different locations within and outside of the plume boundaries. In August 2010, PG&E submitted a Feasibility Study in compliance with Cleanup and Abatement Order (CAO) R6V-2008-0002, evaluating options for comprehensive (Project Area-wide) cleanup of groundwater to background concentrations of chromium. -1- Order No. R6V-2014-0023 3. Project Area and Operable Units The Project Area regulated under this Order is approximately 50 square miles (32,159 acres) in size and includes all areas within the chromium plume boundaries containing more than the maximum background levels of 3.1 micrograms per liter (µg/L) hexavalent chromium or 3.2 µg/L total chromium (based on the Discharger's fourth quarter 2012 groundwater monitoring report) and approximately 1 mile beyond. -
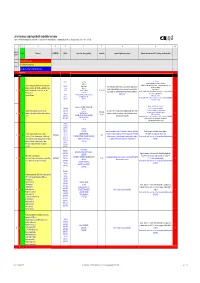
List of Substances Subject to Prohibited / Undesirable / Declaration
List of substances subject to prohibited / undesirable / declaration Appendix 1 to the Guidelines to Design and Purchasing Prohibition of Harmful Substances : ebmpapstMulfingen: 90-001 ebmpapstSt.Georgen: WN 3-12.2 ebmpapstLandshut: 60118.00040 (Ver. 5.0-31.07.2008) 1 2 3 4 5 6 7 8 9 10 Consecu N - new Substance EU-INDEX-No.: CAS-No. Source (law, decree, guideline). Hazard/risk example of application/occurrence Remarks and comments (of 0.1% deviating consideration limit) tive No. V = prohibited (verboten) U = undesirable (unerwünscht) D = subject to declaration (deklarationspflichtig) V = prohibited RoHS from 100 ppm 7439-92-1 TRGS 905, Pb-stabilisers and pigments are subject to declaration. GefStoffV, Prohibited: Anhydrite neutral lead carbonate, lead hydrogen carbonate and lead Lead and compounds (red lead oxide, lead sulphate, lead (7446-14-2 ChemVerbotsV, cable coating, heat transfer medium for accumulators, hybrid integrated sulphate as dye pigment hydrogen carbonate, lead carbonate, lead phtalates, lead 1319-46-6 BatterieV, circuits, stabilisers in plastics, vessels and pipes for aggressive liquids, < 0.4 % in batteries 1 V N acetates, lead phosphates, lead stereates , etc) lead 598-63-0 2002/95/EG (RoHS), K3, R 1, R 3 V E F according to RoHS: Lead as alloying additive in aluminum (up to 0.4 mass%) in steel (up to 67/458/EEC pigment production, lead alloys, anti-corrosion agents (fuel additives), chromate: refer to 0.35 mass%), in copper (up to 4%), 301-04-2 76/769/EEC (89/667/EEC, 97/10/EC, 97/56/EC) soldering agent chromium (VI) -

Hexavalent Chromium (Cr VI) in Drinking Water
PUBLIC HEALTH GOALS FOR CHEMICALS IN DRINKING WATER HEXAVALENT CHROMIUM (Cr VI) July 2011 Governor of the State of California Edmund G. Brown Jr. Acting Secretary for Environmental Protection California Environmental Protection Agency Linda S. Adams Acting Director Office of Environmental Health Hazard Assessment George V. Alexeeff, Ph.D. Public Health Goal for Hexavalent Chromium (Cr VI) in Drinking Water Prepared by Pesticide and Environmental Toxicology Branch Office of Environmental Health Hazard Assessment California Environmental Protection Agency July 2011 PREFACE Drinking Water Public Health Goals Pesticide and Environmental Toxicology Branch Office of Environmental Health Hazard Assessment California Environmental Protection Agency This Public Health Goal (PHG) technical support document provides information on health effects from contaminants in drinking water. PHGs are developed for chemical contaminants based on the best available toxicological data in the scientific literature. These documents and the analyses contained in them provide estimates of the levels of contaminants in drinking water that would pose no significant health risk to individuals consuming the water on a daily basis over a lifetime. The California Safe Drinking Water Act of 1996 (Health and Safety Code, Section 116365) requires the Office of Environmental Health Hazard Assessment (OEHHA) to perform risk assessments and adopt PHGs for contaminants in drinking water based exclusively on public health considerations. The Act requires that PHGs be set in accordance with the following criteria: 1. PHGs for acutely toxic substances shall be set at levels at which no known or anticipated adverse effects on health will occur, with an adequate margin of safety. 2. PHGs for carcinogens or other substances that may cause chronic disease shall be based solely on health effects and shall be set at levels that OEHHA has determined do not pose any significant risk to health. -

Massachusetts Chemical Fact Sheet
Massachusetts Chemical Fact Sheet Hexavalent Chromium Table 1: HEXAVALENT CHROMIUM COMPOUNDS: Compounds SELECTED EXAMPLES* Compound Chemical Formula CAS # This fact sheet is part of a series of chemical fact sheets Ammonium chromate (NH ) Cr0 7788-98-9 developed by TURI to help Massachusetts companies, 4 2 4 community organizations and residents understand the Ammonium dichromate (NH4)2Cr2O7 7789-09-5 chemical’s use and health and environmental effects, as Barium chromate BaCrO4 10294-40-3 well as the availability of safer alternatives. tert-Butyl Chromate [(CH3)3CO]2CrO2 1189-85-1 Hexavalent chromium compounds are a toxic form of Calcium chromate CaCrO4 13765-19-0 chromium and are used in a variety of industrial processes Chromic acid H2CrO4 7738-94-5 and products. Chromium VI chloride CrCl6 14986-48-2 Hexavalent chromium compounds are human carcinogens, Chromic trioxide CrO3 1333-82-0 mutagens and developmental toxicants and are acutely Hexavalent chromium ion Cr6+ 18540-29-9 toxic. Non-hexavalent chromium compounds do not pose Lead chromate PbCrO4 7758-97-6 the same level of concern with regard to either chronic or Lead chromate oxide PbCrO4-PbO 8454-12-1 acute toxicity. Potassium chlorochromate KCrO3Cl 16037-50-6 Until 2011, all chromium compounds were treated as Potassium chromate K2CrO4 7789-00-6 a single category under TURA. Beginning with Potassium dichromate K Cr O 7778-50-9 reporting year 2012, hexavalent chromium 2 2 7 compounds are reportable under TURA as a Silver chromate Ag2CrO4 7784-01-2 separate category and are designated as a Higher Sodium chromate Na2CrO4 7775-11-3 Hazard Substance, which lowers the reporting Sodium dichromate 7789-12-0 threshold to 1,000 lb/year. -

Sd0000029 Evaluation of Chromfte Ore and The
SD0000029 EVALUATION OF CHROMFTE ORE AND THE OPTIMUM METHODS FOR INDUSTRIAL EXTRACTION OF CHROMIUM A TI1HSIS SUBMITTED BY Bakheit Mustafa Mohamed Salih IN CANDIDATURE FOR TIU:DI:GRFJ:OI MASTER OF SC1HNCT DEPARTMENT OF CHEMISTRY FACULTY OF SCIENCE (P.O.BOX 321) OCTOBER 1999 UNIVERSITY OF KHARTOUM 31/ 28 ABSTRACT Samples of chromite ore, collected from Gam and Cheikay mining area (Ingessana Hills) in east Sudan, were analysed to assess the chromium content. Methods for extraction and analysis of chromium metal were developed and established. Analysis were carried out using atomic absorption spectroscopy (AAS) to estimate the contents of chromium, iron, calcium, and magnesium. X-ray fluorescence (XRF) was used to evaluate the levels of chromium, iron, and calcium in the ore. Volumetric analysis was performed to assess chromium and iron, whilst gravimetric analysis was employed to measure the amounts of calcium, magnesium, aluminum and silicon present in the ore. The data was chemically and statistically analyzed to compare the results obtained by the given analytical methods. The results are in good agreement except iron oxide, which displayed a significantly different value when measured by x-ray fluorescence. The data obtained exhibited similarity in almost all cases, when compared with local and global researches, reports, and literature. The study has revealed the average contents of Cr2O3, FeO, CaO, MgO, A12O3, and SiO2 as 40.66. 11.96, 11.94. 0.36. 16.94, 11.45% respectively. MnO and NiO were detected in trace amounts, the corresponding levels in the ore being 72 and 27 ppm. The average chromium content in extracted potassium dichromate measured by using AAS, XRF, and volumetric methods was found to be 31.7%. -

Potassium Dichromate CAS Number: 7778-50-9 EC Number: 231-906-6
21.5.2010 COMMENTS AND RESPONSE TO COMMENTS ON ANNEX XV SVHC: PROPOSAL AND JUSTIFICATION Substance name: Potassium dichromate CAS number: 7778-50-9 EC number: 231-906-6 Reason of the submission of the Annex XV: CMR Disclaimer: The European Chemicals Agency is not responsible for the content of this document. The Response to Comments table has been prepared by the competent authority of the Member State preparing the proposal for identification of a Substance of Very High Concern. The comments were received during the public consultation of the Annex XV dossier. General comments Date Submitted by (name, Comment Response Organisation/MSCA) 20100323 CLEAPSS, UK (on behalf of an CLEAPSS is an advisory service providing support in No answer expected. organisation) science and technology for a consortium of local authorities and their schools including establishments for pupils with special needs. Independent schools, post-16 colleges, teacher training establishments, curriculum developers and others can apply for associate membership. We offer help from nursery education through to A-level studies or equivalent. Our services cover health & safety, risk assessment, sources and use of chemicals, living organisms and equipment. CLEAPSS also provides advice on technicians & their jobs, as well as the design of laboratories and facilities & fittings for D&T and science rooms. 20100324 Germany, company (on behalf of an We reject the ban of Potassium Dichromate in general, Thank for your comment. organisation) because the substance is used in a low concentration (< 1%) in an almost closed system of cuvette tests 20100408 Draeger Safety AG & Co. KGaA, comments k2Cr2O7R1.doc Thank you for this information.