A Quantitative 1H NMR Method for Screening Cannabinoids in CBD Oils
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cannabinoids in the Pathophysiology of Skin Inflammation
molecules Review Cannabinoids in the Pathophysiology of Skin Inflammation Cristian Scheau 1 , Ioana Anca Badarau 1, Livia-Gratiela Mihai 1, Andreea-Elena Scheau 2, Daniel Octavian Costache 3, Carolina Constantin 4,5, Daniela Calina 6 , Constantin Caruntu 1,7,*, Raluca Simona Costache 8,* and Ana Caruntu 9,10 1 Department of Physiology, “Carol Davila” University of Medicine and Pharmacy, 050474 Bucharest, Romania; [email protected] (C.S.); [email protected] (I.A.B.); [email protected] (L.-G.M.) 2 Department of Radiology and Medical Imaging, Fundeni Clinical Institute, 022328 Bucharest, Romania; [email protected] 3 Department of Dermatology, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 4 Immunology Department, ”Victor Babes” National Institute of Pathology, 050096 Bucharest, Romania; [email protected] 5 Department of Pathology, Colentina University Hospital, 020125 Bucharest, Romania 6 Department of Clinical Pharmacy, University of Medicine and Pharmacy of Craiova, 200349 Craiova, Romania; [email protected] 7 Department of Dermatology, “Prof. N. Paulescu” National Institute of Diabetes, Nutrition and Metabolic Diseases, 011233 Bucharest, Romania 8 Gastroenterology and Internal Medicine Clinic, Carol Davila University Central Emergency Military Hospital, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania 9 Department of Oral and Maxillofacial Surgery, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 10 Faculty of Medicine, “Titu Maiorescu” University, 031593 Bucharest, Romania * Correspondence: [email protected] (C.C.); [email protected] (R.S.C.); Tel.: +40-745-086-978 (C.C.) Academic Editor: Eric J. Downer Received: 30 December 2019; Accepted: 2 February 2020; Published: 4 February 2020 Abstract: Cannabinoids are increasingly-used substances in the treatment of chronic pain, some neuropsychiatric disorders and more recently, skin disorders with an inflammatory component. -
Marinol Cannabidiol C21H30O2 Trade Name
Cannabinoids are a group of terpenophenolic compounds secreted by Cannabis flowers that provide relief from a wide array of symptoms including, pain, nausea, and inflammation. They operate by imitating the body’s natural endocannabinoids, which activate to maintain internal stability and overall health. When consumed, cannabinoids bind to receptor sites throughout the brain (CB1 receptors) and body (CB2 receptors). Different cannabinoids have different effects based on their binding affinity for each receptor. By targeting specific cannabinoids at these receptors, different types of relief can be achieved. Presently, there are at least 113 different cannabinoids isolated from Cannabis—each exhibiting varied effects. THC Tetrahydrocannabinol C21H30O2 Trade name: Marinol Legal Status: US – Schedule I, Schedule II (as Cesamet), Schedule III (as Marinol) OH CA – Schedule II UK – Class B AU – S8 (controlled) H Psychoactive Tetrahydrocannabinol (THC) is typically the most abundant cannabinoid present in cannabis products on the market today. THC has very high psychoactive characteristics and is associated with the ‘high’ and euphoria experienced when using cannabis products. When smoked or ingested, THC binds to cannabinoid receptors throughout the body and affects memory, O coordination, concentration, pleasure, and time perception. H Medicinal Benefits Analgesic • Anti-nauseant • Appetite Stimulant Reduces Glaucoma Symptoms Sleep Aid • Reduces Anxiety and PTSD Symptoms CBD Cannabidiol C21H30O2 Trade name: Epidiolex OH Legal Status: US – Schedule I CA – Schedule II UK – POM (Perscription only) H AU – S4 (Perscription only) non-psychoactive Cannabidiol (CBD) is a major phytocannabinoid and accounts for up to 40% of the plant’s extract. Due to its lack of psychoactivity and HO non-interference with motor and psychological functions, it is a leading candidate for a wide variety of medical applications. -

Recent Developments in Cannabis Chemistry
Recent Developments in Cannabis Chemistry BY ALEXANDER T. SHULGIN, Ph.D. The marijuana plant Cannabis sativa contains a bewildering Introduction array of organic chemicals. As is true with other botanic species, there are representatives of almost all chemical classes present, including mono- and sesquiterpenes, carbohy- drates, aromatics, and a variety of nitrogenous compounds. Interest in the study of this plant has centered primarily on the resinous fraction, as it is this material that is invested with the pharmacological activity that is peculiar to the plant. This resin is secreted by the female plant as a protective agent during seed ripening, although it can be found as a microscopic exudate through the aerial portions of plants of either sex. The pure resin, hashish or charas, is the most potent fraction of the plant, and has served as the source material for most of the chemical studies. The family of chemicals that has been isolated from this source has been referred to as the cannabinoid group. It is unique amongst psychotropic materials from plants in that there are no alkaloids present. The fraction is totally nitro- gen-free. Rather, the set of compounds can be considered as analogs of the parent compound cannabinol (I), a fusion product of terpene and a substituted resorcinol. Beyond the scope of this present review are such questions as the distribution of these compounds within the plant, the bo- tanic variability resulting from geographic distribution, the diversity of pharmacological action assignable to the several Reprinted from Journal of Psychedelic Drugs, vol. II, no. 1, 197 1. 397 398 Marijuana: Medical Papers distinct compounds present, and the various preparations and customs of administration. -

Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry
Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry Dedication To Steve Örebro Studies in Chemistry 19 NICOLE RIDDELL Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry © Nicole Riddell, 2017 Title: Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry Publisher: Örebro University 2017 www.publications.oru.se Print: Örebro University, Repro 04/2017 ISSN 1651-4270 ISBN 978-91-7529-184-0 Abstract Nicole Riddell (2017): Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry. Örebro Studies in Chemistry 19. Although gas and liquid chromatography have emerged as dominant separation techniques in environmental analytical chemistry, these methods do not allow for the concurrent analysis of chemically diverse groups of persistent organic pollutants (POPs). There are also a small number of compounds which are not easily amenable to either of these traditional separation techniques. The main objective of this thesis was to address these issues by demonstrating the applicability of packed col- umn supercritical fluid chromatography (pSFC) coupled to mass spec- trometry (MS) in various aspects of environmental chemistry. First, pSFC/MS analytical methods were developed for legacy POPs (PCDDs, PCDFs, and PCBs) as well as the emerging environmental con- taminant Dechlorane Plus (DP), and issues relating to the ionization of target analytes when pSFC was coupled to MS were explored. Novel APPI and APCI reagents (fluorobenzene and triethylamine) were opti- mized and real samples (water and soil) were analyzed to demonstrate environmental applicability. The possibility of chiral and preparative scale pSFC separations was then demonstrated through the isolation and characterization of ther- mally labile hexabromocyclododecane (HBCDD) stereoisomers. The analytical pSFC separation of the α-, β-, and γ-HBCDD enantiomers as well as the δ and ε meso forms was shown to be superior to results ob- tained using a published LC method. -

Deuteriodesilylation: a Mild and Selective Method for the Site- Specific Incorporation of Deuterium Into Drug Candidates and Pharmaceutical Structures
DEUTERIODESILYLATION: A MILD AND SELECTIVE METHOD FOR THE SITE- SPECIFIC INCORPORATION OF DEUTERIUM INTO DRUG CANDIDATES AND PHARMACEUTICAL STRUCTURES Kimberly N. Voronin A Thesis Submitted to the University of North Carolina Wilmington in Partial Fulfillment of the Requirements for the Degree of Master of Science Department of Chemistry and Biochemistry University of North Carolina Wilmington 2012 Approved by Advisory Committee Chris V. Galliford Pamela J. Seaton John A. Tyrell Chair Accepted by Dean, Graduate School TABLE OF CONTENTS ABSTRACT ................................................................................................................................... vi ACKNOWLEDGEMENTS .......................................................................................................... vii DEDICATION ............................................................................................................................... ix LIST OF TABLES .......................................................................................................................... x LIST OF FIGURES ...................................................................................................................... xi LIST OF ABBREVIATIONS ...................................................................................................... xiii LIST OF SCHEMES .....................................................................................................................xv CHAPTER 1: INTRODUCTION ....................................................................................................1 -

INTRODUCTION One of the Tasks of a Pharmacologist Is the Classification of Drugs
NEUROPSYCHOPHARMACOLOGIC STUDIES OF MARIJUANA: SOME SYNTHETIC AND NATURAL THC DERIVATIVES IN ANIMALS AND MAN* Edward F. Domino Department of Pharmacology, University of Michigan, Ann Arbor 48104 and Lafayette Clinic, Detroit, Mich. 48207 INTRODUCTION One of the tasks of a pharmacologist is the classification of drugs. Marijuana and its derivatives pose some problems. Legally marijuana has been classified as a narcotic, yet pharmacologically it is quite different. Mankind has known about the Cannabis plant from which marijuana and other products are obtained for more than 4,708 years. A considerable body of data is available to describe its gross effects. Over the centuries it has been called a depressant, euphoriant, ine- briant, intoxicant, hallucinogen, psychotomimetic, sedative-hypnotic-anesthetic, and stimulant. Perhaps more than any other drug it is a social irritant that elicits rather remarkable behavior reactions in both users and nonusers. Moreau’ in 1845, employed it to produce a model psychosis, thus initiating the fashion to study hallucinogens as a means of gaining insights into mental illness. Lewin2 classified Cannabis as one of the “phantastica: hallucinating substances.” One text on hallucinogens3 does not even refer to Cannabis, but another does? There are some aspects of the subjective effects of both LSD-25 and Cannabis that are similar, yet the latter produces sedative effects, no significant sympatho- mimetic actions, and no cross-tolerance to LSD-25. Hollister5 reviewed the re- cent findings on the effects in man of marijuana and its putative active ingredient Ag-THC, stressing that current investigators have, for the most part, confirmed the Cannabis-induced clinical syndromes long known to occur. -

The Use of Cannabinoids in Animals and Therapeutic Implications for Veterinary Medicine: a Review
Veterinarni Medicina, 61, 2016 (3): 111–122 Review Article doi: 10.17221/8762-VETMED The use of cannabinoids in animals and therapeutic implications for veterinary medicine: a review L. Landa1, A. Sulcova2, P. Gbelec3 1Faculty of Medicine, Masaryk University, Brno, Czech Republic 2Central European Institute of Technology, Masaryk University, Brno, Czech Republic 3Veterinary Hospital and Ambulance AA Vet, Prague, Czech Republic ABSTRACT: Cannabinoids/medical marijuana and their possible therapeutic use have received increased atten- tion in human medicine during the last years. This increased attention is also an issue for veterinarians because particularly companion animal owners now show an increased interest in the use of these compounds in veteri- nary medicine. This review sets out to comprehensively summarise well known facts concerning properties of cannabinoids, their mechanisms of action, role of cannabinoid receptors and their classification. It outlines the main pharmacological effects of cannabinoids in laboratory rodents and it also discusses examples of possible beneficial use in other animal species (ferrets, cats, dogs, monkeys) that have been reported in the scientific lit- erature. Finally, the article deals with the prospective use of cannabinoids in veterinary medicine. We have not intended to review the topic of cannabinoids in an exhaustive manner; rather, our aim was to provide both the scientific community and clinical veterinarians with a brief, concise and understandable overview of the use of cannabinoids in veterinary -

Interferometric Observations of Warm Deuterated Methanol in the Inner Regions of Low- Mass Protostars
Interferometric observations of warm deuterated methanol in the inner regions of low- mass protostars Taquet, V; Bianchi, E.; Codella, C.; Persson, M., V; Ceccarelli, C.; Cabrit, S.; Jorgensen, J. K.; Kahane, C.; Lopez-Sepulcre, A.; Neri, R. Published in: Astronomy & Astrophysics DOI: 10.1051/0004-6361/201936044 Publication date: 2019 Document version Publisher's PDF, also known as Version of record Document license: CC BY-NC Citation for published version (APA): Taquet, V., Bianchi, E., Codella, C., Persson, M. V., Ceccarelli, C., Cabrit, S., Jorgensen, J. K., Kahane, C., Lopez-Sepulcre, A., & Neri, R. (2019). Interferometric observations of warm deuterated methanol in the inner regions of low-mass protostars. Astronomy & Astrophysics, 632, [A19]. https://doi.org/10.1051/0004- 6361/201936044 Download date: 02. Oct. 2021 A&A 632, A19 (2019) https://doi.org/10.1051/0004-6361/201936044 Astronomy & © ESO 2019 Astrophysics Interferometric observations of warm deuterated methanol in the inner regions of low-mass protostars? V. Taquet1, E. Bianchi2,1, C. Codella1,2, M. V. Persson3, C. Ceccarelli2, S. Cabrit4, J. K. Jørgensen5, C. Kahane2, A. López-Sepulcre2,6, and R. Neri6 1 INAF, Osservatorio Astrofisico di Arcetri, Largo E. Fermi 5, 50125 Firenze, Italy e-mail: [email protected] 2 IPAG, Université Grenoble Alpes, CNRS, 38000 Grenoble, France 3 Department of Space, Earth, and Environment, Chalmers University of Technology, Onsala Space Observatory, 439 92 Onsala, Sweden 4 LERMA, Observatoire de Paris, PSL Research University, CNRS, Sorbonne Université, UPMC Université Paris 06, 75014 Paris, France 5 Niels Bohr Institute, University of Copenhagen, Øster Voldgade 5–7, 1350 Copenhagen K., Denmark 6 Institut de Radioastronomie Millimétrique, 38406 Saint-Martin d’Hères, France Received 7 June 2019 / Accepted 19 September 2019 ABSTRACT Methanol is a key species in astrochemistry because it is the most abundant organic molecule in the interstellar medium and is thought to be the mother molecule of many complex organic species. -

Methanol—Inhibitor Or Promoter of the Formation of Gas Hydrates from Deuterated Ice?
BOBEV AND TAIT: METHANOL—INHIBITOR OR PROMOTER? 1209 American Mineralogist, Volume 89, pages 1208–1214, 2004 Methanol—inhibitor or promoter of the formation of gas hydrates from deuterated ice? SVILEN BOBEV* AND KIMBERLY T. TAIT Manuel Lujan Jr. Neutron Scattering Center, LANSCE 12 MS H805, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, U.S.A. ABSTRACT Kinetic studies are reported of the effect of methanol on the rate of formation of CO2- and CH4-hy- drates by means of in situ time-of-flight neutron powder diffraction. The experiments were carried out at temperatures ranging from 200 to 250 K and pressures up to 7 MPa. The samples were prepared from mixtures of ground, deuterated ice and deuterated methanol (up to 20 vol%), which were transformed in situ into CO2- or CH4-hydrates by pressurizing the systems with the corresponding gas. The observed rates of formation of hydrates are orders of magnitude higher than the rate of formation from pure deuterated ice under the same pressure and temperature conditions. Glycols and alcohols, methanol in particular, are long known as thermodynamic inhibitors of hydrate formation. Our study indicates that methanol can also act as a kinetic promoter for the formation of gas hydrates. Preliminary data suggest that the kinetics also depend strongly on concentration and the isotopic composition. INTRODUCTION particular, time-dependent neutron diffraction at a variety of tem- The crystal structures, thermodynamic models, and engineer- peratures and pressures has been effectively used to research the ing applications of CO2- and CH4-hydrates have been extensively kinetics of gas hydrate formation and dissociation (Henning et al. -

Hydrogen Isotope Exchanges Between Water and Methanol in Interstellar Ices a Faure, M Faure, P Theulé, E Quirico, B Schmitt
Hydrogen isotope exchanges between water and methanol in interstellar ices A Faure, M Faure, P Theulé, E Quirico, B Schmitt To cite this version: A Faure, M Faure, P Theulé, E Quirico, B Schmitt. Hydrogen isotope exchanges between water and methanol in interstellar ices. Astronomy and Astrophysics - A&A, EDP Sciences, 2015, 584, pp.A98. 10.1051/0004-6361/201526499. hal-01452286 HAL Id: hal-01452286 https://hal.archives-ouvertes.fr/hal-01452286 Submitted on 1 Feb 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. A&A 584, A98 (2015) Astronomy DOI: 10.1051/0004-6361/201526499 & c ESO 2015 Astrophysics Hydrogen isotope exchanges between water and methanol in interstellar ices A. Faure1,2,M.Faure1,2, P. Theulé3, E. Quirico1,2, and B. Schmitt1,2 1 Univ. Grenoble Alpes, IPAG, 38000 Grenoble, France e-mail: [email protected] 2 CNRS, IPAG, 38000 Grenoble, France 3 Aix-Marseille Université, PIIM UMR-CNRS 7345, 13397 Marseille, France Received 8 May 2015 / Accepted 22 September 2015 ABSTRACT The deuterium fractionation of gas-phase molecules in hot cores is believed to reflect the composition of interstellar ices. -

The Effects of Cannabinoids on Stress-Coping
Utah State University DigitalCommons@USU All Graduate Theses and Dissertations Graduate Studies 5-2021 The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredtictable Stress: A Preclinical Systematic Review and Meta-Analysis Noa Reuveni Utah State University Follow this and additional works at: https://digitalcommons.usu.edu/etd Part of the Psychology Commons Recommended Citation Reuveni, Noa, "The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredtictable Stress: A Preclinical Systematic Review and Meta-Analysis" (2021). All Graduate Theses and Dissertations. 8098. https://digitalcommons.usu.edu/etd/8098 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@USU. It has been accepted for inclusion in All Graduate Theses and Dissertations by an authorized administrator of DigitalCommons@USU. For more information, please contact [email protected]. THE EFFECTS OF CANNABINOIDS ON STRESS-COPING BEHAVIORS AND NEUROENDOCRINOLOGICAL MEASURES IN CHRONIC UNPREDTICTABLE STRESS: A PRECLINICAL SYSTEMATIC REVIEW AND META-ANALYSIS by Noa Reuveni A thesis submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Psychology Approved: __________________ __________________ Sara Freeman, Ph.D. Scott Bates, Ph.D. Co-Major Professor Co-Major Professor __________________ __________________ Diana Meter, Ph.D. Sarah Schwartz, Ph.D. Committee Member Committee Member __________________ __________________ Tyler Renshaw, Ph.D. D. Richard Cutler, Ph.D. Committee Member Interim Vice Provost for Graduate Studies UTAH STATE UNIVERSITY Logan, Utah 2021 ii Copyright © Noa Reuveni 2021 All Rights Reserved iii ABSTRACT The Effects of Cannabinoids on Stress-Coping Behaviors and Neuroendocrinological Measures in Chronic Unpredictable Stress: A Preclinical Systematic Review and Meta-Analysis by Noa Reuveni, Master of Science Utah State University, 2021 Major Professors: Dr. -
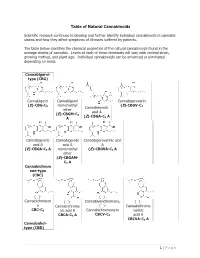
Table of Natural Cannabinoids
Table of Natural Cannabinoids Scientific research continues to develop and further identify individual cannabinoids in cannabis strains and how they affect symptoms of illnesses suffered by patients. The table below identifies the chemical properties of the natural cannabinoids found in the average strains of cannabis. Levels of each of these chemicals will vary with varietal strain, growing method, and plant age. Individual cannabinoids can be enhanced or eliminated depending on need. Cannabigerol- type (CBG) Cannabigerol Cannabigerol Cannabigerovarin (E)-CBG-C monomethyl (E)-CBGV-C 5 Cannabinerolic 3 ether acid A (E)-CBGM-C 5 (Z)-CBGA-C A A 5 Cannabigerolic Cannabigerolic Cannabigerovarinic acid acid A acid A A (E)-CBGA-C5 A monomethyl (E)-CBGVA-C3 A ether (E)-CBGAM- C5 A Cannabichrom ene-type (CBC) (±)- (±)- Cannabichromen (±)- Cannabivarichromene, (±)- e Cannabichrome (±)- Cannabichrome CBC-C5 nic acid A Cannabichromevarin varinic CBCA-C5 A CBCV-C3 acid A CBCVA-C3 A Cannabidiol- type (CBD) 1 | Page (−)-Cannabidiol Cannabidiol Cannabidiol-C4 (−)- Cannabidiorc CBD-C5 momomethyl CBD-C4 Cannabidivarin ol ether CBDV-C3 CBD-C1 CBDM-C5 Cannabidiolic Cannabidivarini acid c acid CBDA-C5 CBDVA-C3 Cannabinodiol- type (CBND) Cannabinodiol Cannabinodivar CBND-C5 in CBND-C3 Tetrahydrocan nabinol-type (THC) 9 9 9 Δ - Δ - Δ - Δ9- Tetrahydrocanna Tetrahydrocan Tetrahydrocannabivarin 9 Tetrahydrocan binol nabinol-C4 Δ -THCV-C3 9 9 nabiorcol Δ -THC-C5 Δ -THC-C4 9 Δ -THCO-C1 9 9 Δ -Tetrahydro- Δ9-Tetrahydro- Δ -Tetrahydro- Δ9-Tetrahydro- cannabinolic