INFORMATION to USERS the Most Advanced Technology Has Been Used to Photo Graph and Reproduce This Manuscript from the Microfilm Master
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cannabinoids in the Pathophysiology of Skin Inflammation
molecules Review Cannabinoids in the Pathophysiology of Skin Inflammation Cristian Scheau 1 , Ioana Anca Badarau 1, Livia-Gratiela Mihai 1, Andreea-Elena Scheau 2, Daniel Octavian Costache 3, Carolina Constantin 4,5, Daniela Calina 6 , Constantin Caruntu 1,7,*, Raluca Simona Costache 8,* and Ana Caruntu 9,10 1 Department of Physiology, “Carol Davila” University of Medicine and Pharmacy, 050474 Bucharest, Romania; [email protected] (C.S.); [email protected] (I.A.B.); [email protected] (L.-G.M.) 2 Department of Radiology and Medical Imaging, Fundeni Clinical Institute, 022328 Bucharest, Romania; [email protected] 3 Department of Dermatology, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 4 Immunology Department, ”Victor Babes” National Institute of Pathology, 050096 Bucharest, Romania; [email protected] 5 Department of Pathology, Colentina University Hospital, 020125 Bucharest, Romania 6 Department of Clinical Pharmacy, University of Medicine and Pharmacy of Craiova, 200349 Craiova, Romania; [email protected] 7 Department of Dermatology, “Prof. N. Paulescu” National Institute of Diabetes, Nutrition and Metabolic Diseases, 011233 Bucharest, Romania 8 Gastroenterology and Internal Medicine Clinic, Carol Davila University Central Emergency Military Hospital, Carol Davila University of Medicine and Pharmacy, 050474 Bucharest, Romania 9 Department of Oral and Maxillofacial Surgery, “Carol Davila” Central Military Emergency Hospital, 010825 Bucharest, Romania; [email protected] 10 Faculty of Medicine, “Titu Maiorescu” University, 031593 Bucharest, Romania * Correspondence: [email protected] (C.C.); [email protected] (R.S.C.); Tel.: +40-745-086-978 (C.C.) Academic Editor: Eric J. Downer Received: 30 December 2019; Accepted: 2 February 2020; Published: 4 February 2020 Abstract: Cannabinoids are increasingly-used substances in the treatment of chronic pain, some neuropsychiatric disorders and more recently, skin disorders with an inflammatory component. -

Nabilone for Chronic Pain Management: a Review of Clinical Effectiveness, Safety, and Guidelines
TITLE: Nabilone for Chronic Pain Management: A Review of Clinical Effectiveness, Safety, and Guidelines DATE: 11 November 2011 CONTEXT AND POLICY ISSUES Cannabis sativa is a flowering plant that has long been used as a recreational drug, and for medicinal purposes.1,2 The main psychoactive component of cannabis is delta-9- tetrahydrocannabinol (∆9-THC).2 Nabilone (Cesamet®) is an oral synthetic cannabinoid, which is licensed in Canada for treating patients with severe nausea and vomiting related to chemotherapy for cancer and who have failed to respond adequately to conventional antiemetic treatments.2-4 Clinical trials and anecdotal reports have suggested that the use of nabilone in other medical conditions, such as appetite stimulation, anxiety, spasticity, and pain.1,5 Chronic pain affects approximately one in five people in developed countries and two in five in less well-resourced countries. In many circumstances, the patient’s quality of life is poor due to persistent pain caused either by an ongoing illness or nerve damage caused by the disease after resolution or cure of the disease.6 Multiple sclerosis (MS) is a neurodegenerative disease, and is the most common cause of neurological disability in young people, with an average age of onset around 30 years and a prevalence of about 120 per 100,000 individuals in North America. The majority of patients with MS display symptoms, such as fatigue, muscle stiffness or spasticity, pain, memory problems, balance trouble, tremors, urinary disturbance, and sexual dysfunctions.2,7 The purpose of this review is to assess the evidence of benefits and harms related to the use of nabilone in management of chronic pain, including patients with MS. -

Problems of Drug Dependence 1980 Proceedings of the 42Nd Annual Scientific Meeting the Committee on Problems of Drug Dependence
National Institute on Drug Abuse MONOGRAPH SERIES Problems of Drug Dependence 1980 Proceedings of the 42nd Annual Scientific Meeting The Committee on Problems of Drug Dependence, Inc. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES • Public Health Service • Alcohol, Drug Abuse, and Mental Health Administration Problems of Drug Dependence, 1980 Proceedings of the 42nd Annual Scientific Meeting, The Committee on Problems of Drug Dependence, Inc. Editor: Louis S. Harris, Ph.D. NIDA Research Monograph 34 February 1981 DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service Alcohol, Drug Abuse, and Mental Health Administration National Institute on Drug Abuse Division of Research 5600 Fishers Lane Rockville, Maryland 20857 For sale by the Superintendent of Documents, U.S. Government Printing Office Washington, D.C. 20402 The NIDA Research Monograph series is prepared by the Division of Research of the National Institute on Drug Abuse. Its primary objective is to provide critical reviews of research problem areas and techniques, the content of state-of-the-art conferences, integrative research reviews and significant original research. Its dual publication emphasis is rapid and targeted dissemination to the scientific and professional community. Editorial Advisory Board Avram Goldstein, M.D. Addiction Research Foundation Palo Alto, California Jerome Jaffe, M.D. College of Physicians and Surgeons Columbia University, New York Reese T. Jones, M.D. Langley Porter Neuropsychiatric Institute University of California San Francisco, California William McGlothlin, Ph.D. Deportment of Psychology, UCLA Los Angeles, California Jack Mendelson, M.D. Alcohol and Drug Abuse Research Center Harvard Medical School McLean Hospital Belmont, Massachusetts Helen Nowlis, Ph.D. Office of Drug Education, DHHS Washington, D.C Lee Robins, Ph.D. -
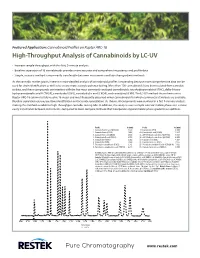
High-Throughput Analysis of Cannabinoids by LC-UV
Featured Application: Cannabinoid Profiles on Raptor ARC-18 High-Throughput Analysis of Cannabinoids by LC-UV • Increase sample throughput with this fast, 9-minute analysis. • Baseline separation of 16 cannabinoids provides more accurate and comprehensive potency and profile data. • Simple, isocratic method is more easily transferable between instruments and labs than gradient methods. As the cannabis market grows, interest in more detailed analysis of cannabinoid profiles is expanding because more comprehensive data can be used for strain identification as well as to ensure more accurate potency testing. More than 100 cannabinoids have been isolated from cannabis to date, and these compounds can interfere with the five most commonly analyzed cannabinoids: tetrahydrocannabinol (THC), delta-9-tetra- hydrocannabinolic acid A (THCA), cannabidiol (CBD), cannabidiolic acid (CBDA), and cannabinol (CBN). The LC-UV method shown here uses a Raptor ARC-18 column to fully resolve 16 major and most frequently observed minor cannabinoids for which commercial standards are available. Baseline separation ensures positive identification and accurate quantitation. As shown, all compounds were resolved in a fast 9-minute analysis, making this method suitable for high-throughput cannabis testing labs. In addition, this analysis uses a simple isocratic mobile phase so it is more easily transferable between instruments, compared to more complex methods that incorporate atypical mobile phase gradients or additives. Peaks tR (min) Peaks tR (min) 1. Cannabidivarinic acid (CBDVA) 1.877 9. Cannabinol (CBN) 4.609 2. Cannabidivarin (CBDV) 2.086 10. Cannabinolic acid (CBNA) 5.437 3. Cannabidiolic acid (CBDA) 2.592 11. Δ9-Tetrahydrocannabinol (Δ9-THC) 5.815 4. Cannabigerolic acid (CBGA) 2.750 12. -

(12) Patent Application Publication (10) Pub. No.: US 2012/0115729 A1 Qin Et Al
US 201201.15729A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2012/0115729 A1 Qin et al. (43) Pub. Date: May 10, 2012 (54) PROCESS FOR FORMING FILMS, FIBERS, Publication Classification AND BEADS FROM CHITNOUS BOMASS (51) Int. Cl (75) Inventors: Ying Qin, Tuscaloosa, AL (US); AOIN 25/00 (2006.01) Robin D. Rogers, Tuscaloosa, AL A6II 47/36 (2006.01) AL(US); (US) Daniel T. Daly, Tuscaloosa, tish 9.8 (2006.01)C (52) U.S. Cl. ............ 504/358:536/20: 514/777; 426/658 (73) Assignee: THE BOARD OF TRUSTEES OF THE UNIVERSITY OF 57 ABSTRACT ALABAMA, Tuscaloosa, AL (US) (57) Disclosed is a process for forming films, fibers, and beads (21) Appl. No.: 13/375,245 comprising a chitinous mass, for example, chitin, chitosan obtained from one or more biomasses. The disclosed process (22) PCT Filed: Jun. 1, 2010 can be used to prepare films, fibers, and beads comprising only polymers, i.e., chitin, obtained from a suitable biomass, (86). PCT No.: PCT/US 10/36904 or the films, fibers, and beads can comprise a mixture of polymers obtained from a suitable biomass and a naturally S3712). (4) (c)(1), Date: Jan. 26, 2012 occurring and/or synthetic polymer. Disclosed herein are the (2), (4) Date: an. AO. films, fibers, and beads obtained from the disclosed process. O O This Abstract is presented solely to aid in searching the sub Related U.S. Application Data ject matter disclosed herein and is not intended to define, (60)60) Provisional applicationpp No. 61/182,833,sy- - - s filed on Jun. -

Potential Health Benefits of Cannabis Extracts: a Review Maria Rosana Ramirez
International Journal of Chemical and Biomedical Science 2016; 2(1): 1-8 Published online March 1, 2016 (http://www.aascit.org/journal/ijcbs) Potential Health Benefits of Cannabis Extracts: A Review Maria Rosana Ramirez CITER-The National Scientific and Technical Research Council (CONICET), Argentine Government, Agency, Argentina Email address [email protected] Citation Maria Rosana Ramirez. Potential Health Benefits of Cannabis Extracts: A Review. International Journal of Chemical and Biomedical Science . Vol. 2, No. 1, 2016, pp. 1-8. Keywords Abstract Non-cannabinoids Compounds, A central tenet underlying the use of plant preparations is that herbs contain many Cannabis Extracts, bioactive compounds. Cannabis contains tetrahydrocannabinols (THC) a primary Bioactivity metabolite with reported psychotropic effects. Therefore, the presence of THC makes controversial the use of Cannabis to treat diseases by which their uses and applications were limited. The question then is: is it possible to use the extracts from Cannabis to treat the diseases related with it use in folk medicine? More recently, the synergistic Received: January 24, 2016 contributions of bioactive constituents have been scientifically demonstrated. We Revised: February 10, 2016 reviewed the literature concerning medical cannabis and its secondary metabolites, Accepted: February 12, 2016 including fraction and total extracts. Scientific evidence shows that secondary metabolites in cannabis may enhance the positive effects of THC a primary metabolite. Other chemical components (cannabinoid and non-cannabinoid) in cannabis or its extracts may reduce THC-induced anxiety, cholinergic deficits, and immunosuppression; which could increase its therapeutic potential. Particular attention will be placed on non- cannabinoid compounds interactions that could produce synergy with respect to treatment of pain, inflammation, epilepsy, fungal and bacterial infections. -

Medicinal Chemistry Endeavors Around the Phytocannabinoids
CHEMISTRY & BIODIVERSITY – Vol. 4 (2007) 1707 REVIEW Medicinal Chemistry Endeavors around the Phytocannabinoids by Eric Stern and Didier M. Lambert* Drug Design and Discovery Center and Unite´ de Chimie pharmaceutique et de Radiopharmacie, Ecole de Pharmacie, Faculte´ de Me´decine, Universite´ catholique de Louvain, Avenue E. Mounier 73, U.C.L. 73.40, B-1200 Bruxelles (phone: þ3227647347; fax: þ3227647363; e-mail: [email protected]) Over the past 50 years, a considerable research in medicinal chemistry has been carried out around the natural constituents of Cannabis sativa L. Following the identification of D9-tetrahydrocannabinol (D9-THC) in 1964, critical chemical modifications, e.g., variation of the side chain at C(3) and the opening of the tricyclic scaffold, have led to the characterization of potent and cannabinoid receptor subtype-selective ligands. Those ligands that demonstrate high affinity for the cannabinoid receptors and good biological efficacy are still used as powerful pharmacological tools. This review summarizes past as well as recent developments in the structure–activity relationships of phytocannabinoids. 1. Introduction. – Despite the wide uses of preparations of the hemp Cannabis sativa L. during the History, the modern pharmacology of natural cannabinoids has been hampered by the slow progress in the elucidations of the chemical structures of its major components. Indeed, it is nowadays known that more than 70 compounds derived from a diterpene structure are present in the plant [1], and this fact may explain the difficulty to obtain pure chemical entities in the past. In addition, the medicinal research for more than a half century has been driven by the search for the components responsible for the psychoactive effects of cannabis, this era in the history of the chemical research on cannabinoids have been recently reviewed [2][3]. -
Marinol Cannabidiol C21H30O2 Trade Name
Cannabinoids are a group of terpenophenolic compounds secreted by Cannabis flowers that provide relief from a wide array of symptoms including, pain, nausea, and inflammation. They operate by imitating the body’s natural endocannabinoids, which activate to maintain internal stability and overall health. When consumed, cannabinoids bind to receptor sites throughout the brain (CB1 receptors) and body (CB2 receptors). Different cannabinoids have different effects based on their binding affinity for each receptor. By targeting specific cannabinoids at these receptors, different types of relief can be achieved. Presently, there are at least 113 different cannabinoids isolated from Cannabis—each exhibiting varied effects. THC Tetrahydrocannabinol C21H30O2 Trade name: Marinol Legal Status: US – Schedule I, Schedule II (as Cesamet), Schedule III (as Marinol) OH CA – Schedule II UK – Class B AU – S8 (controlled) H Psychoactive Tetrahydrocannabinol (THC) is typically the most abundant cannabinoid present in cannabis products on the market today. THC has very high psychoactive characteristics and is associated with the ‘high’ and euphoria experienced when using cannabis products. When smoked or ingested, THC binds to cannabinoid receptors throughout the body and affects memory, O coordination, concentration, pleasure, and time perception. H Medicinal Benefits Analgesic • Anti-nauseant • Appetite Stimulant Reduces Glaucoma Symptoms Sleep Aid • Reduces Anxiety and PTSD Symptoms CBD Cannabidiol C21H30O2 Trade name: Epidiolex OH Legal Status: US – Schedule I CA – Schedule II UK – POM (Perscription only) H AU – S4 (Perscription only) non-psychoactive Cannabidiol (CBD) is a major phytocannabinoid and accounts for up to 40% of the plant’s extract. Due to its lack of psychoactivity and HO non-interference with motor and psychological functions, it is a leading candidate for a wide variety of medical applications. -

Recent Developments in Cannabis Chemistry
Recent Developments in Cannabis Chemistry BY ALEXANDER T. SHULGIN, Ph.D. The marijuana plant Cannabis sativa contains a bewildering Introduction array of organic chemicals. As is true with other botanic species, there are representatives of almost all chemical classes present, including mono- and sesquiterpenes, carbohy- drates, aromatics, and a variety of nitrogenous compounds. Interest in the study of this plant has centered primarily on the resinous fraction, as it is this material that is invested with the pharmacological activity that is peculiar to the plant. This resin is secreted by the female plant as a protective agent during seed ripening, although it can be found as a microscopic exudate through the aerial portions of plants of either sex. The pure resin, hashish or charas, is the most potent fraction of the plant, and has served as the source material for most of the chemical studies. The family of chemicals that has been isolated from this source has been referred to as the cannabinoid group. It is unique amongst psychotropic materials from plants in that there are no alkaloids present. The fraction is totally nitro- gen-free. Rather, the set of compounds can be considered as analogs of the parent compound cannabinol (I), a fusion product of terpene and a substituted resorcinol. Beyond the scope of this present review are such questions as the distribution of these compounds within the plant, the bo- tanic variability resulting from geographic distribution, the diversity of pharmacological action assignable to the several Reprinted from Journal of Psychedelic Drugs, vol. II, no. 1, 197 1. 397 398 Marijuana: Medical Papers distinct compounds present, and the various preparations and customs of administration. -

Article 22 Regulation for Restriction of Synthetic Drugs
ARTICLE 22 REGULATION FOR RESTRICTION OF SYNTHETIC DRUGS SECTION 22.1 AUTHORITY This regulation is promulgated under the authority granted to the Needham Board of Health under Massachusetts General Laws Chapter 111, Section 31 which states that “boards of health may make reasonable health regulations”. SECTION 22.2 PURPOSE The Needham Board of Health has found that synthetic marijuana, consisting of plant or other material treated with various chemicals or other synthetic substances not approved for human consumption, may be marketed and sold as herbal incense in the greater Boston area, although they are being used in the same manner and for the same purposes as scheduled drugs. In addition, the use of these products has become particularly popular among teens and young adults. Based on information and reports from hospitals, emergency room doctors, and police agencies, individuals who use these products experience dangerous side effects including convulsions, hallucinations, and dangerously elevated heart rates. This is evidence that synthetic marijuana products are harmful if inhaled or consumed, and present a significant public health danger. These synthetic compounds and others have a high potential for abuse and lack of any accepted medical use, these dangerous products, while not approved for human consumption, are marketed and sold in a form that allows for such consumption, putting at risk the individuals who come into contact with them. Therefore, the Needham Board of Health adopts this regulation for the purpose and with the intent to protect the public health and safety of the Town of Needham and its residents from the threat posed by the availability and use of synthetic marijuana, synthetic stimulants, synthetic hallucinogens, and other dangerous products by prohibiting persons from trafficking in, possessing, and using them within the town. -

Federal Register/Vol. 84, No. 214/Tuesday, November 5, 2019
Federal Register / Vol. 84, No. 214 / Tuesday, November 5, 2019 / Notices 59645 Authority: 42 U.S.C. 6213; and 30 CFR company plans to bulk manufacture Controlled substance Drug Schedule 556.511–556.515. these drugs as synthetics. No other code Walter D. Cruickshank, activities for these drug codes are Hydromorphone ....................... 9150 II Acting Director, Bureau of Ocean Energy authorized for this registration. Hydrocodone ............................ 9193 II Management. Morphine .................................. 9300 II Dated: October 18, 2019. Oripavine .................................. 9330 II [FR Doc. 2019–24052 Filed 11–4–19; 8:45 am] William T. McDermott, Thebaine .................................. 9333 II BILLING CODE 4310–MR–P Assistant Administrator. Opium extracts ......................... 9610 II Opium fluid extract ................... 9620 II [FR Doc. 2019–24107 Filed 11–4–19; 8:45 am] Opium tincture ......................... 9630 II BILLING CODE 4410–09–P Opium, powdered .................... 9639 II DEPARTMENT OF JUSTICE Opium, granulated ................... 9640 II Oxymorphone .......................... 9652 II Drug Enforcement Administration Noroxymorphone ..................... 9668 II DEPARTMENT OF JUSTICE Tapentadol ............................... 9780 II [Docket No. DEA–536] Drug Enforcement Administration The company plans to manufacture Bulk Manufacturer of Controlled [Docket No. DEA–526] the listed controlled substances as an Substances Application: Organix, Inc. Active Pharmaceutical Ingredient (API) Bulk -

2 Spice English Presentation
Spice Spice contains no compensatory substances Специи не содержит компенсационные вещества Spice is a mix of herbs (shredded plant material) and manmade chemicals with mind-altering effects. It is often called “synthetic marijuana” because some of the chemicals in it are similar to ones in marijuana; but its effects are sometimes very different from marijuana, and frequently much stronger. It is most often labeled “Not for Human Consumption” and disguised as incense. Eliminationprocess • The synthetic agonists such as THC is fat soluble. • Probably, they are stored as THC in cell membranes. • Some of the chemicals in Spice, however, attach to those receptors more strongly than THC, which could lead to a much stronger and more unpredictable effect. • Additionally, there are many chemicals that remain unidentified in products sold as Spice and it is therefore not clear how they may affect the user. • Moreover, these chemicals are often being changed as the makers of Spice alter them to avoid the products being illegal. • To dissolve the Spice crystals Acetone is used endocannabinoids synhtetic THC cannabinoids CB1 and CB2 agonister Binds to cannabinoidreceptor CB1 CB2 - In the brain -in the immune system Decreased avtivity in the cell ____________________ Maria Ellgren Since some of the compounds have a longer toxic effects compared to naturally THC, as reported: • negative effects that often occur the day after consumption, as a general hangover , but without nausea, mentally slow, confused, distracted, impairment of long and short term memory • Other reports mention the qualitative impairment of cognitive processes and emotional functioning, like all the oxygen leaves the brain.