Supplementary Materials
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
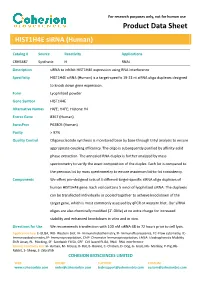
Product Data Sheet
For research purposes only, not for human use Product Data Sheet HIST1H4E siRNA (Human) Catalog # Source Reactivity Applications CRH5487 Synthetic H RNAi Description siRNA to inhibit HIST1H4E expression using RNA interference Specificity HIST1H4E siRNA (Human) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol HIST1H4E Alternative Names H4/E; H4FE; Histone H4 Entrez Gene 8367 (Human) SwissProt P62805 (Human) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of human HIST1H4E gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, IH- Immunohistochemistry, -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers
Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers writer effector eg. HAT, HMT reader eg. bromodomain eraser eg. HDAC, KDM The writer HAT1: A known H4 lysine 5,12 di-acetyltransferase writer siHAT1 siControl HAT1 H4 K12Ac H4 K5Ac actin Western blot for histone H4 modifications after control and HAT1 siRNA transfections. HAT1: EGF-stimulated immunoprecipitation specific specific - - HAT1 IgG HAT1 IgG - - R α non R α non EGF: + + + - - - WB: HAT1 Immunoprecipitation / WB to measure HAT1 levels +/- Heatmap of gene expression changes of all human histone EGF acetyltransferases +/- EGF and siRNA treatments shows HAT1 expression is EGF-dependent Working model of HAT1 : The oldest “new” histone acetyltransferase EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase Surprise: HAT1 also binds (a few sites) on chromatin HAT1 ChipSeq signal sits on Hist1 locus on Chromosome 6 Read Depth HAT1 bound sites (zoom) HAT1 ChIP-seq peaks cluster at Read Depth histone H4 promoters. Hist1H2BE Hist1H4D Hist1H3D Hist1H4E Is HAT1 a transcription factor for its substrate (H4)? EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase HAT1 is required for S-phase burst of histone H4 mRNA HAT1 Hist1H4B mRNA level 50 Rbap46 45 shCont-3 H4 40 shHAT1-A7 35 shHAT1-B6 30 25 B6 A7 - - 20 15 shHAT1 shHAT1 shControl 10 Gene actin)Expression (versus 5 HAT1 0 0 2 4 6 8 10 actin hours after release from double thymidine block G1 S G2/M HAT1 loss: Life with less histones EGF -

Genome-Wide Association Study of Dietary Intake in the UK Biobank Study and Its Associations with Schizophrenia and Other Traits Maria Niarchou1,2,3, Enda M
Niarchou et al. Translational Psychiatry (2020) 10:51 https://doi.org/10.1038/s41398-020-0688-y Translational Psychiatry ARTICLE Open Access Genome-wide association study of dietary intake in the UK biobank study and its associations with schizophrenia and other traits Maria Niarchou1,2,3, Enda M. Byrne1, Maciej Trzaskowski4, Julia Sidorenko 1,5, Kathryn E. Kemper1, John J. McGrath 6,7,8,MichaelC.O’ Donovan 2,MichaelJ.Owen2 and Naomi R. Wray 1,6 Abstract Motivated by observational studies that report associations between schizophrenia and traits, such as poor diet, increased body mass index and metabolic disease, we investigated the genetic contribution to dietary intake in a sample of 335,576 individuals from the UK Biobank study. A principal component analysis applied to diet question item responses generated two components: Diet Component 1 (DC1) represented a meat-related diet and Diet Component 2 (DC2) a fish and plant-related diet. Genome-wide association analysis identified 29 independent single- nucleotide polymorphisms (SNPs) associated with DC1 and 63 SNPs with DC2. Estimated from over 35,000 3rd-degree relative pairs that are unlikely to share close family environments, heritabilities for both DC1 and DC2 were 0.16 (standard error (s.e.) = 0.05). SNP-based heritability was 0.06 (s.e. = 0.003) for DC1 and 0.08 (s.e = 0.004) for DC2. We estimated significant genetic correlations between both DCs and schizophrenia, and several other traits. Mendelian randomisation analyses indicated a negative uni-directional relationship between liability to schizophrenia and tendency towards selecting a meat-based diet (which could be direct or via unidentified correlated variables), but a bi- fi 1234567890():,; 1234567890():,; 1234567890():,; 1234567890():,; directional relationship between liability to schizophrenia and tendency towards selecting a sh and plant-based diet consistent with genetic pleiotropy. -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

MYC-Containing Amplicons in Acute Myeloid Leukemia: Genomic Structures, Evolution, and Transcriptional Consequences
Leukemia (2018) 32:2152–2166 https://doi.org/10.1038/s41375-018-0033-0 ARTICLE Acute myeloid leukemia Corrected: Correction MYC-containing amplicons in acute myeloid leukemia: genomic structures, evolution, and transcriptional consequences 1 1 2 2 1 Alberto L’Abbate ● Doron Tolomeo ● Ingrid Cifola ● Marco Severgnini ● Antonella Turchiano ● 3 3 1 1 1 Bartolomeo Augello ● Gabriella Squeo ● Pietro D’Addabbo ● Debora Traversa ● Giulia Daniele ● 1 1 3 3 4 Angelo Lonoce ● Mariella Pafundi ● Massimo Carella ● Orazio Palumbo ● Anna Dolnik ● 5 5 6 2 7 Dominique Muehlematter ● Jacqueline Schoumans ● Nadine Van Roy ● Gianluca De Bellis ● Giovanni Martinelli ● 3 4 8 1 Giuseppe Merla ● Lars Bullinger ● Claudia Haferlach ● Clelia Tiziana Storlazzi Received: 4 August 2017 / Revised: 27 October 2017 / Accepted: 13 November 2017 / Published online: 22 February 2018 © The Author(s) 2018. This article is published with open access Abstract Double minutes (dmin), homogeneously staining regions, and ring chromosomes are vehicles of gene amplification in cancer. The underlying mechanism leading to their formation as well as their structure and function in acute myeloid leukemia (AML) remain mysterious. We combined a range of high-resolution genomic methods to investigate the architecture and expression pattern of amplicons involving chromosome band 8q24 in 23 cases of AML (AML-amp). This 1234567890();,: revealed that different MYC-dmin architectures can coexist within the same leukemic cell population, indicating a step-wise evolution rather than a single event origin, such as through chromothripsis. This was supported also by the analysis of the chromothripsis criteria, that poorly matched the model in our samples. Furthermore, we found that dmin could evolve toward ring chromosomes stabilized by neocentromeres. -

(12) United States Patent (10) Patent No.: US 7,799,528 B2 Civin Et Al
US007799528B2 (12) United States Patent (10) Patent No.: US 7,799,528 B2 Civin et al. (45) Date of Patent: Sep. 21, 2010 (54) THERAPEUTIC AND DIAGNOSTIC Al-Hajj et al., “Prospective identification of tumorigenic breast can APPLICATIONS OF GENES cer cells.” Proc. Natl. Acad. Sci. U.S.A., 100(7):3983-3988 (2003). DIFFERENTIALLY EXPRESSED IN Bhatia et al., “A newly discovered class of human hematopoietic cells LYMPHO-HEMATOPOETC STEM CELLS with SCID-repopulating activity.” Nat. Med. 4(9)1038-45 (1998). (75) Inventors: Curt I. Civin, Baltimore, MD (US); Bonnet, D., “Normal and leukemic CD35-negative human Robert W. Georgantas, III, Towson, hematopoletic stem cells.” Rev. Clin. Exp. Hematol. 5:42-61 (2001). Cambot et al., “Human Immune Associated Nucleotide 1: a member MD (US) of a new guanosine triphosphatase family expressed in resting T and (73) Assignee: The Johns Hopkins University, B cells.” Blood, 99(9):3293-3301 (2002). Baltimore, MD (US) Chen et al., “Kruppel-like Factor 4 (Gut-enriched Kruppel-like Fac tor) Inhibits Cell Proliferation by Blocking G1/S Progression of the *) NotOt1Ce: Subjubject to anyy d1Sclaimer,disclai theh term off thisthi Cell Cycle.” J. Biol. Chem., 276(32):30423-30428 (2001). patent is extended or adjusted under 35 Chen et al., “Transcriptional profiling of Kurppel-like factor 4 reveals U.S.C. 154(b) by 168 days. a function in cell cycle regulation and epithelial differentiation.” J. Mol. Biol. 326(3):665-677 (2003). (21) Appl. No.: 11/199.665 Civin et al., “Highly purified CD34-positive cells reconstitute (22) Filed: Aug. 9, 2005 hematopoiesis,” J. -

Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B
Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B by Geoffrey Graham Lundell-Smith A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Biochemistry University of Toronto © Copyright by Geoffrey Lundell-Smith, 2017 Characterizing Novel Interactions of Transcriptional Repression Proteins BCL6 and BCL6B Geoffrey Graham Lundell-Smith Masters of Science Department of Biochemistry University of Toronto 2016 Abstract B-cell Lymphoma 6 (BCL6) and its close homolog BCL6B encode proteins that are members of the BTB-Zinc Finger family of transcription factors. BCL6 plays an important role in regulating the differentiation and proliferation of B-cells during the adaptive immune response, and is also involved in T cell development and inflammation. BCL6 acts by repressing genes involved in DNA damage response during the affinity maturation of immunoglobulins, and the mis- expression of BCL6 can lead to diffuse large B-cell lymphoma. Although BCL6B shares high sequence similarity with BCL6, the functions of BCL6B are not well-characterized. I used BioID, an in vivo proximity-dependent labeling method, to identify novel BCL6 and BCL6B protein interactors and validated a number of these interactions with co-purification experiments. I also examined the evolutionary relationship between BCL6 and BCL6B and identified conserved residues in an important interaction interface that mediates corepressor binding and gene repression. ii Acknowledgments Thank you to my supervisor, Gil Privé for his mentorship, guidance, and advice, and for giving me the opportunity to work in his lab. Thanks to my committee members, Dr. John Rubinstein and Dr. Jeff Lee for their ideas, thoughts, and feedback during my Masters. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

Candidate DNA Methylation Drivers of Acquired Cisplatin Resistance in Ovarian Cancer Identified by Methylome and Expression Profiling
Oncogene (2012) 31, 4567–4576 & 2012 Macmillan Publishers Limited All rights reserved 0950-9232/12 www.nature.com/onc ONCOGENOMICS Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling C Zeller1, W Dai1, NL Steele2, A Siddiq3, AJ Walley3, CSM Wilhelm-Benartzi1, S Rizzo4, A van der Zee5, JA Plumb6 and R Brown1,4 1Epigenetics Unit, Department of Surgery and Cancer, Imperial College London, London, UK; 2Beatson West of Scotland Cancer Centre, Glasgow, UK; 3Department of Genomics of Common Disease, School of Public Health, Hammersmith Hospital, London, UK; 4Section of Medicine, Institute of Cancer Research, Sutton, UK; 5Department of Obstetrics and Gynecology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands and 6Cancer Research UK Beatson Laboratories, Institute of Cancer Sciences, University of Glasgow, Glasgow, UK Multiple DNA methylation changes in the cancer Keywords: drug resistance; cisplatin; DNA methylation; methylome are associated with the acquisition of drug ovarian cancer; DNMTi; HDACi resistance; however it remains uncertain how many represent critical DNA methylation drivers of chemore- sistance. Using isogenic, cisplatin-sensitive/resistant ovar- ian cancer cell lines and inducing resensitizaton with demethylating agents, we aimed to identify consistent Introduction methylation and expression changes associated with chemoresistance. Using genome-wide DNA methylation Ovarian cancer often presents at an advanced stage with profiling across 27 578 CpG sites, we identified loci at limited chances for curative treatment (Ozols, 2004). 4092 genes becoming hypermethylated in chemoresistant Common treatment strategies include debulking surgery A2780/cp70 compared with the parental-sensitive A2780 in combination with chemotherapy, which usually cell line.