GSTT1 Copy Number Gain and ZNF Overexpression Are Predictors of Poor Response to Imatinib in Gastrointestinal Stromal Tumors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
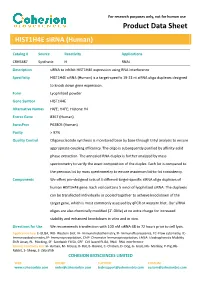
Product Data Sheet
For research purposes only, not for human use Product Data Sheet HIST1H4E siRNA (Human) Catalog # Source Reactivity Applications CRH5487 Synthetic H RNAi Description siRNA to inhibit HIST1H4E expression using RNA interference Specificity HIST1H4E siRNA (Human) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol HIST1H4E Alternative Names H4/E; H4FE; Histone H4 Entrez Gene 8367 (Human) SwissProt P62805 (Human) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of human HIST1H4E gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, IH- Immunohistochemistry, -

Autism Multiplex Family with 16P11.2P12.2 Microduplication Syndrome in Monozygotic Twins and Distal 16P11.2 Deletion in Their Brother
European Journal of Human Genetics (2012) 20, 540–546 & 2012 Macmillan Publishers Limited All rights reserved 1018-4813/12 www.nature.com/ejhg ARTICLE Autism multiplex family with 16p11.2p12.2 microduplication syndrome in monozygotic twins and distal 16p11.2 deletion in their brother Anne-Claude Tabet1,2,3,4, Marion Pilorge2,3,4, Richard Delorme5,6,Fre´de´rique Amsellem5,6, Jean-Marc Pinard7, Marion Leboyer6,8,9, Alain Verloes10, Brigitte Benzacken1,11,12 and Catalina Betancur*,2,3,4 The pericentromeric region of chromosome 16p is rich in segmental duplications that predispose to rearrangements through non-allelic homologous recombination. Several recurrent copy number variations have been described recently in chromosome 16p. 16p11.2 rearrangements (29.5–30.1 Mb) are associated with autism, intellectual disability (ID) and other neurodevelopmental disorders. Another recognizable but less common microdeletion syndrome in 16p11.2p12.2 (21.4 to 28.5–30.1 Mb) has been described in six individuals with ID, whereas apparently reciprocal duplications, studied by standard cytogenetic and fluorescence in situ hybridization techniques, have been reported in three patients with autism spectrum disorders. Here, we report a multiplex family with three boys affected with autism, including two monozygotic twins carrying a de novo 16p11.2p12.2 duplication of 8.95 Mb (21.28–30.23 Mb) characterized by single-nucleotide polymorphism array, encompassing both the 16p11.2 and 16p11.2p12.2 regions. The twins exhibited autism, severe ID, and dysmorphic features, including a triangular face, deep-set eyes, large and prominent nasal bridge, and tall, slender build. The eldest brother presented with autism, mild ID, early-onset obesity and normal craniofacial features, and carried a smaller, overlapping 16p11.2 microdeletion of 847 kb (28.40–29.25 Mb), inherited from his apparently healthy father. -

Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers
Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers writer effector eg. HAT, HMT reader eg. bromodomain eraser eg. HDAC, KDM The writer HAT1: A known H4 lysine 5,12 di-acetyltransferase writer siHAT1 siControl HAT1 H4 K12Ac H4 K5Ac actin Western blot for histone H4 modifications after control and HAT1 siRNA transfections. HAT1: EGF-stimulated immunoprecipitation specific specific - - HAT1 IgG HAT1 IgG - - R α non R α non EGF: + + + - - - WB: HAT1 Immunoprecipitation / WB to measure HAT1 levels +/- Heatmap of gene expression changes of all human histone EGF acetyltransferases +/- EGF and siRNA treatments shows HAT1 expression is EGF-dependent Working model of HAT1 : The oldest “new” histone acetyltransferase EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase Surprise: HAT1 also binds (a few sites) on chromatin HAT1 ChipSeq signal sits on Hist1 locus on Chromosome 6 Read Depth HAT1 bound sites (zoom) HAT1 ChIP-seq peaks cluster at Read Depth histone H4 promoters. Hist1H2BE Hist1H4D Hist1H3D Hist1H4E Is HAT1 a transcription factor for its substrate (H4)? EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase HAT1 is required for S-phase burst of histone H4 mRNA HAT1 Hist1H4B mRNA level 50 Rbap46 45 shCont-3 H4 40 shHAT1-A7 35 shHAT1-B6 30 25 B6 A7 - - 20 15 shHAT1 shHAT1 shControl 10 Gene actin)Expression (versus 5 HAT1 0 0 2 4 6 8 10 actin hours after release from double thymidine block G1 S G2/M HAT1 loss: Life with less histones EGF -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B
Characterizing Novel Interactions of Transcriptional Repressor Proteins BCL6 & BCL6B by Geoffrey Graham Lundell-Smith A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Biochemistry University of Toronto © Copyright by Geoffrey Lundell-Smith, 2017 Characterizing Novel Interactions of Transcriptional Repression Proteins BCL6 and BCL6B Geoffrey Graham Lundell-Smith Masters of Science Department of Biochemistry University of Toronto 2016 Abstract B-cell Lymphoma 6 (BCL6) and its close homolog BCL6B encode proteins that are members of the BTB-Zinc Finger family of transcription factors. BCL6 plays an important role in regulating the differentiation and proliferation of B-cells during the adaptive immune response, and is also involved in T cell development and inflammation. BCL6 acts by repressing genes involved in DNA damage response during the affinity maturation of immunoglobulins, and the mis- expression of BCL6 can lead to diffuse large B-cell lymphoma. Although BCL6B shares high sequence similarity with BCL6, the functions of BCL6B are not well-characterized. I used BioID, an in vivo proximity-dependent labeling method, to identify novel BCL6 and BCL6B protein interactors and validated a number of these interactions with co-purification experiments. I also examined the evolutionary relationship between BCL6 and BCL6B and identified conserved residues in an important interaction interface that mediates corepressor binding and gene repression. ii Acknowledgments Thank you to my supervisor, Gil Privé for his mentorship, guidance, and advice, and for giving me the opportunity to work in his lab. Thanks to my committee members, Dr. John Rubinstein and Dr. Jeff Lee for their ideas, thoughts, and feedback during my Masters. -

Signature Redacted Thesis Supervisor Certified By
Single-Cell Transcriptomics of the Mouse Thalamic Reticular Nucleus by Taibo Li S.B., Massachusetts Institute of Technology (2015) Submitted to the Department of Electrical Engineering and Computer Science in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science at the MASSACHUSETTS INSTITUTE OF TECHNOLOGY June 2017 @ Massachusetts Institute of Technology 2017. All rights reserved. A uthor ... ..................... Department of Electrical Engineering and Computer Science May 25, 2017 Certified by. 3ignature redacted Guoping Feng Poitras Professor of Neuroscience, MIT Signature redacted Thesis Supervisor Certified by... Kasper Lage Assistant Professor, Harvard Medical School Thesis Supervisor Accepted by . Signature redacted Christopher Terman Chairman, Masters of Engineering Thesis Committee MASSACHUSETTS INSTITUTE 0) OF TECHNOLOGY w AUG 14 2017 LIBRARIES 2 Single-Cell Transcriptomics of the Mouse Thalamic Reticular Nucleus by Taibo Li Submitted to the Department of Electrical Engineering and Computer Science on May 25, 2017, in partial fulfillment of the requirements for the degree of Master of Engineering in Electrical Engineering and Computer Science Abstract The thalamic reticular nucleus (TRN) is strategically located at the interface between the cortex and the thalamus, and plays a key role in regulating thalamo-cortical in- teractions. Current understanding of TRN neurobiology has been limited due to the lack of a comprehensive survey of TRN heterogeneity. In this thesis, I developed an integrative computational framework to analyze the single-nucleus RNA sequencing data of mouse TRN in a data-driven manner. By combining transcriptomic, genetic, and functional proteomic data, I discovered novel insights into the molecular mecha- nisms through which TRN regulates sensory gating, and suggested targeted follow-up experiments to validate these findings. -

Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2019 Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Shufei Song University of Pennsylvania Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Biochemistry Commons, and the Cell Biology Commons Recommended Citation Song, Shufei, "Rap1-Mediated Chromatin And Gene Expression Changes At Senescence" (2019). Publicly Accessible Penn Dissertations. 3557. https://repository.upenn.edu/edissertations/3557 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/3557 For more information, please contact [email protected]. Rap1-Mediated Chromatin And Gene Expression Changes At Senescence Abstract ABSTRACT RAP1-MEDIATED CHROMATIN AND GENE EXPRESSION CHANGES AT SENESCENCE The telomeric protein Rap1 has been extensively studied for its roles as a transcriptional activator and repressor. Indeed, in both yeast and mammals, Rap1 is known to bind throughout the genome to reorganize chromatin and regulate gene transcription. Previously, our lab published evidence that Rap1 plays important roles in cellular senescence. In telomerase-deficient S. cerevisiae, Rap1 relocalizes from telomeres and subtelomeres to new Rap1 target at senescence (NRTS). This leads to two types of histone loss: Rap1 lowers global histone levels by repressing histone gene transcription and it also results in local nucleosome displacement at the promoters of the activated NRTS. Here, I examine mechanisms of site-specific histone loss by presenting evidence that Rap1 can directly interact with histone tetramers H3/H4, and map this interaction to a three-amino-acid-patch within the DNA binding domain. Functional studies are performed in vivo using a mutant form of Rap1 with weakened histone interactions, and deficient promoter clearance as well as blunted gene activation is observed, indicating that direct Rap1-H3/H4 interactions are involved in nucleosome displacement. -

Clinical Impact of Copy Number Variation Changes in Bladder Cancer Samples
EXPERIMENTAL AND THERAPEUTIC MEDICINE 22: 901, 2021 Clinical impact of copy number variation changes in bladder cancer samples VICTORIA SPASOVA1, BORIS MLADENOV2, SIMEON RANGELOV3, ZORA HAMMOUDEH1, DESISLAVA NESHEVA1, DIMITAR SERBEZOV1, RADA STANEVA1,4, SAVINA HADJIDEKOVA1,4, MIHAIL GANEV1, LUBOMIR BALABANSKI1,5, RADOSLAVA VAZHAROVA5,6, CHAVDAR SLAVOV3, DRAGA TONCHEVA1 and OLGA ANTONOVA1 1Department of Medical Genetics, Medical University‑Sofia, 1431 Sofia;2 Department of Urology, UMBALSM N.I. Pirogov, 1606 Sofia; 3Department of Urology, Tsaritsa Yoanna University Hospital, 1527 Sofia; 4Medical Genetics Laboratory, Nadezhda Women's Health Hospital, 1373 Sofia; 5Medical Genetics Laboratory, GARH Malinov, 1680 Sofia; 6Department of Biology, Medical Genetics and Microbiology, Faculty of Medicine, Sofia University St. Kliment Ohridski, 1407 Sofia, Bulgaria Received November 30, 2019; Accepted February 18, 2021 DOI: 10.3892/etm.2021.10333 Abstract. The aim of the present study was to detect copy uroepithelial tumours may lay a foundation for implementing number variations (CNVs) related to tumour progression and molecular CNV profiling of bladder tumours as part of a metastasis of urothelial carcinoma through whole‑genome routine progression risk estimation strategy, thus expanding scanning. A total of 30 bladder cancer samples staged from the personalized therapeutic approach. pTa to pT4 were included in the study. DNA was extracted from freshly frozen tissue via standard phenol‑chloroform extraction Introduction and CNV analysis was performed on two alternative platforms (CytoChip Oligo aCGH, 4x44K and Infinium OncoArray‑500K The most successful approach to treating a disease has BeadChip; Illumina, Inc.). Data were analysed with BlueFuse always been etiological therapy. In the case of bladder Multi software and Karyostudio, respectively. -

Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders
177 Arch Neuropsychitry 2020;57:177−191 RESEARCH ARTICLE https://doi.org/10.29399/npa.24890 Chromosomal Microarray Analysis in Turkish Patients with Unexplained Developmental Delay and Intellectual Developmental Disorders Hakan GÜRKAN1 , Emine İkbal ATLI1 , Engin ATLI1 , Leyla BOZATLI2 , Mengühan ARAZ ALTAY2 , Sinem YALÇINTEPE1 , Yasemin ÖZEN1 , Damla EKER1 , Çisem AKURUT1 , Selma DEMİR1 , Işık GÖRKER2 1Faculty of Medicine, Department of Medical Genetics, Edirne, Trakya University, Edirne, Turkey 2Faculty of Medicine, Department of Child and Adolescent Psychiatry, Trakya University, Edirne, Turkey ABSTRACT Introduction: Aneuploids, copy number variations (CNVs), and single in 39 (39/123=31.7%) patients. Twelve CNV variant of unknown nucleotide variants in specific genes are the main genetic causes of significance (VUS) (9.75%) patients and 7 CNV benign (5.69%) patients developmental delay (DD) and intellectual disability disorder (IDD). were reported. In 6 patients, one or more pathogenic CNVs were These genetic changes can be detected using chromosome analysis, determined. Therefore, the diagnostic efficiency of CMA was found to chromosomal microarray (CMA), and next-generation DNA sequencing be 31.7% (39/123). techniques. Therefore; In this study, we aimed to investigate the Conclusion: Today, genetic analysis is still not part of the routine in the importance of CMA in determining the genomic etiology of unexplained evaluation of IDD patients who present to psychiatry clinics. A genetic DD and IDD in 123 patients. diagnosis from CMA can eliminate genetic question marks and thus Method: For 123 patients, chromosome analysis, DNA fragment analysis alter the clinical management of patients. Approximately one-third and microarray were performed. Conventional G-band karyotype of the positive CMA findings are clinically intervenable. -

University of California, San Diego
UC San Diego UC San Diego Electronic Theses and Dissertations Title The post-terminal differentiation fate of RNAs revealed by next-generation sequencing Permalink https://escholarship.org/uc/item/7324r1rj Author Lefkowitz, Gloria Kuo Publication Date 2012 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs -

Gene Expression in the Mouse Eye: an Online Resource for Genetics Using 103 Strains of Mice
Molecular Vision 2009; 15:1730-1763 <http://www.molvis.org/molvis/v15/a185> © 2009 Molecular Vision Received 3 September 2008 | Accepted 25 August 2009 | Published 31 August 2009 Gene expression in the mouse eye: an online resource for genetics using 103 strains of mice Eldon E. Geisert,1 Lu Lu,2 Natalie E. Freeman-Anderson,1 Justin P. Templeton,1 Mohamed Nassr,1 Xusheng Wang,2 Weikuan Gu,3 Yan Jiao,3 Robert W. Williams2 (First two authors contributed equally to this work) 1Department of Ophthalmology and Center for Vision Research, Memphis, TN; 2Department of Anatomy and Neurobiology and Center for Integrative and Translational Genomics, Memphis, TN; 3Department of Orthopedics, University of Tennessee Health Science Center, Memphis, TN Purpose: Individual differences in patterns of gene expression account for much of the diversity of ocular phenotypes and variation in disease risk. We examined the causes of expression differences, and in their linkage to sequence variants, functional differences, and ocular pathophysiology. Methods: mRNAs from young adult eyes were hybridized to oligomer microarrays (Affymetrix M430v2). Data were embedded in GeneNetwork with millions of single nucleotide polymorphisms, custom array annotation, and information on complementary cellular, functional, and behavioral traits. The data include male and female samples from 28 common strains, 68 BXD recombinant inbred lines, as well as several mutants and knockouts. Results: We provide a fully integrated resource to map, graph, analyze, and test causes and correlations of differences in gene expression in the eye. Covariance in mRNA expression can be used to infer gene function, extract signatures for different cells or tissues, to define molecular networks, and to map quantitative trait loci that produce expression differences.