Rap1-Mediated Chromatin and Gene Expression Changes at Senescence
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
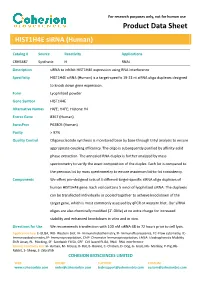
Product Data Sheet
For research purposes only, not for human use Product Data Sheet HIST1H4E siRNA (Human) Catalog # Source Reactivity Applications CRH5487 Synthetic H RNAi Description siRNA to inhibit HIST1H4E expression using RNA interference Specificity HIST1H4E siRNA (Human) is a target-specific 19-23 nt siRNA oligo duplexes designed to knock down gene expression. Form Lyophilized powder Gene Symbol HIST1H4E Alternative Names H4/E; H4FE; Histone H4 Entrez Gene 8367 (Human) SwissProt P62805 (Human) Purity > 97% Quality Control Oligonucleotide synthesis is monitored base by base through trityl analysis to ensure appropriate coupling efficiency. The oligo is subsequently purified by affinity-solid phase extraction. The annealed RNA duplex is further analyzed by mass spectrometry to verify the exact composition of the duplex. Each lot is compared to the previous lot by mass spectrometry to ensure maximum lot-to-lot consistency. Components We offers pre-designed sets of 3 different target-specific siRNA oligo duplexes of human HIST1H4E gene. Each vial contains 5 nmol of lyophilized siRNA. The duplexes can be transfected individually or pooled together to achieve knockdown of the target gene, which is most commonly assessed by qPCR or western blot. Our siRNA oligos are also chemically modified (2’-OMe) at no extra charge for increased stability and enhanced knockdown in vitro and in vivo. Directions for Use We recommends transfection with 100 nM siRNA 48 to 72 hours prior to cell lysis. Application key: E- ELISA, WB- Western blot, IH- Immunohistochemistry, -

Histone Isoform H2A1H Promotes Attainment of Distinct Physiological
Bhattacharya et al. Epigenetics & Chromatin (2017) 10:48 DOI 10.1186/s13072-017-0155-z Epigenetics & Chromatin RESEARCH Open Access Histone isoform H2A1H promotes attainment of distinct physiological states by altering chromatin dynamics Saikat Bhattacharya1,4,6, Divya Reddy1,4, Vinod Jani5†, Nikhil Gadewal3†, Sanket Shah1,4, Raja Reddy2,4, Kakoli Bose2,4, Uddhavesh Sonavane5, Rajendra Joshi5 and Sanjay Gupta1,4* Abstract Background: The distinct functional efects of the replication-dependent histone H2A isoforms have been dem- onstrated; however, the mechanistic basis of the non-redundancy remains unclear. Here, we have investigated the specifc functional contribution of the histone H2A isoform H2A1H, which difers from another isoform H2A2A3 in the identity of only three amino acids. Results: H2A1H exhibits varied expression levels in diferent normal tissues and human cancer cell lines (H2A1C in humans). It also promotes cell proliferation in a context-dependent manner when exogenously overexpressed. To uncover the molecular basis of the non-redundancy, equilibrium unfolding of recombinant H2A1H-H2B dimer was performed. We found that the M51L alteration at the H2A–H2B dimer interface decreases the temperature of melting of H2A1H-H2B by ~ 3 °C as compared to the H2A2A3-H2B dimer. This diference in the dimer stability is also refected in the chromatin dynamics as H2A1H-containing nucleosomes are more stable owing to M51L and K99R substitu- tions. Molecular dynamic simulations suggest that these substitutions increase the number of hydrogen bonds and hydrophobic interactions of H2A1H, enabling it to form more stable nucleosomes. Conclusion: We show that the M51L and K99R substitutions, besides altering the stability of histone–histone and histone–DNA complexes, have the most prominent efect on cell proliferation, suggesting that the nucleosome sta- bility is intimately linked with the physiological efects observed. -

Analysis of Trans Esnps Infers Regulatory Network Architecture
Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2014 © 2014 Anat Kreimer All rights reserved ABSTRACT Analysis of trans eSNPs infers regulatory network architecture Anat Kreimer eSNPs are genetic variants associated with transcript expression levels. The characteristics of such variants highlight their importance and present a unique opportunity for studying gene regulation. eSNPs affect most genes and their cell type specificity can shed light on different processes that are activated in each cell. They can identify functional variants by connecting SNPs that are implicated in disease to a molecular mechanism. Examining eSNPs that are associated with distal genes can provide insights regarding the inference of regulatory networks but also presents challenges due to the high statistical burden of multiple testing. Such association studies allow: simultaneous investigation of many gene expression phenotypes without assuming any prior knowledge and identification of unknown regulators of gene expression while uncovering directionality. This thesis will focus on such distal eSNPs to map regulatory interactions between different loci and expose the architecture of the regulatory network defined by such interactions. We develop novel computational approaches and apply them to genetics-genomics data in human. We go beyond pairwise interactions to define network motifs, including regulatory modules and bi-fan structures, showing them to be prevalent in real data and exposing distinct attributes of such arrangements. We project eSNP associations onto a protein-protein interaction network to expose topological properties of eSNPs and their targets and highlight different modes of distal regulation. -

Table S1. 49 Histone Variants Were Identified with High Sequence Coverage Through LC-MS/MS Analysis Electronic Supplementary
Electronic Supplementary Material (ESI) for Analytical Methods. This journal is © The Royal Society of Chemistry 2020 Table S1. 49 histone variants were identified with high sequence coverage through LC-MS/MS analysis Sequence Uniprot IDs Protein Name Protein Description Coverage Ratio E2+/E2- RSD P07305 H10_HUMAN 67.5% Histone H1.0 OS=Homo sapiens GN=H1F0 PE=1 SV=3 4.85 23.3% Histone H1.1 OS=Homo sapiens GN=HIST1H1A PE=1 Q02539 H11_HUMAN 74.4% SV=3 0.35 92.6% Histone H1.2 OS=Homo sapiens GN=HIST1H1C PE=1 P16403 H12_HUMAN 67.1% SV=2 0.73 80.6% Histone H1.3 OS=Homo sapiens GN=HIST1H1D PE=1 P16402 H13_HUMAN 63.8% SV=2 0.75 77.7% Histone H1.4 OS=Homo sapiens GN=HIST1H1E PE=1 P10412 H14_HUMAN 69.0% SV=2 0.70 80.3% Histone H1.5 OS=Homo sapiens GN=HIST1H1B PE=1 P16401 H15_HUMAN 79.6% SV=3 0.29 98.3% Testis-specific H1 histone OS=Homo sapiens GN=H1FNT Q75WM6 H1FNT_HUMAN 7.8% \ \ PE=2 SV=3 Histone H1oo OS=Homo sapiens GN=H1FOO PE=2 Q8IZA3 H1FOO_HUMAN 5.2% \ \ SV=1 Histone H1t OS=Homo sapiens GN=HIST1H1T PE=2 P22492 H1T_HUMAN 31.4% SV=4 1.42 65.0% Q92522 H1X_HUMAN 82.6% Histone H1x OS=Homo sapiens GN=H1FX PE=1 SV=1 1.15 33.2% Histone H2A type 1 OS=Homo sapiens GN=HIST1H2AG P0C0S8 H2A1_HUMAN 99.2% PE=1 SV=2 0.57 26.8% Q96QV6 H2A1A_HUMAN 58.0% Histone H2A type 1-A OS=Homo sapiens 0.90 11.2% GN=HIST1H2AA PE=1 SV=3 Histone H2A type 1-B/E OS=Homo sapiens P04908 H2A1B_HUMAN 99.2% GN=HIST1H2AB PE=1 SV=2 0.92 30.2% Histone H2A type 1-C OS=Homo sapiens Q93077 H2A1C_HUMAN 100.0% GN=HIST1H2AC PE=1 SV=3 0.76 27.6% Histone H2A type 1-D OS=Homo sapiens P20671 -

A Cell Line P53 Mutation Type UM
A Cell line p53 mutation Type UM-SCC 1 wt UM-SCC5 Exon 5, 157 GTC --> TTC Missense mutation by transversion (Valine --> Phenylalanine UM-SCC6 wt UM-SCC9 wt UM-SCC11A wt UM-SCC11B Exon 7, 242 TGC --> TCC Missense mutation by transversion (Cysteine --> Serine) UM-SCC22A Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC22B Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC38 Exon 5, 132 AAG --> AAT Missense mutation by transversion (Lysine --> Asparagine) UM-SCC46 Exon 8, 278 CCT --> CGT Missense mutation by transversion (Proline --> Alanine) B 1 Supplementary Methods Cell Lines and Cell Culture A panel of ten established HNSCC cell lines from the University of Michigan series (UM-SCC) was obtained from Dr. T. E. Carey at the University of Michigan, Ann Arbor, MI. The UM-SCC cell lines were derived from eight patients with SCC of the upper aerodigestive tract (supplemental Table 1). Patient age at tumor diagnosis ranged from 37 to 72 years. The cell lines selected were obtained from patients with stage I-IV tumors, distributed among oral, pharyngeal and laryngeal sites. All the patients had aggressive disease, with early recurrence and death within two years of therapy. Cell lines established from single isolates of a patient specimen are designated by a numeric designation, and where isolates from two time points or anatomical sites were obtained, the designation includes an alphabetical suffix (i.e., "A" or "B"). The cell lines were maintained in Eagle's minimal essential media supplemented with 10% fetal bovine serum and penicillin/streptomycin. -

Genome-Wide Approach to Identify Risk Factors for Therapy-Related Myeloid Leukemia
Leukemia (2006) 20, 239–246 & 2006 Nature Publishing Group All rights reserved 0887-6924/06 $30.00 www.nature.com/leu ORIGINAL ARTICLE Genome-wide approach to identify risk factors for therapy-related myeloid leukemia A Bogni1, C Cheng2, W Liu2, W Yang1, J Pfeffer1, S Mukatira3, D French1, JR Downing4, C-H Pui4,5,6 and MV Relling1,6 1Department of Pharmaceutical Sciences, The University of Tennessee, Memphis, TN, USA; 2Department of Biostatistics, The University of Tennessee, Memphis, TN, USA; 3Hartwell Center, The University of Tennessee, Memphis, TN, USA; 4Department of Pathology, The University of Tennessee, Memphis, TN, USA; 5Department of Hematology/Oncology St Jude Children’s Research Hospital, The University of Tennessee, Memphis, TN, USA; and 6Colleges of Medicine and Pharmacy, The University of Tennessee, Memphis, TN, USA Using a target gene approach, only a few host genetic risk therapy increases, the importance of identifying host factors for factors for treatment-related myeloid leukemia (t-ML) have been secondary neoplasms increases. defined. Gene expression microarrays allow for a more 4 genome-wide approach to assess possible genetic risk factors Because DNA microarrays interrogate multiple ( 10 000) for t-ML. We assessed gene expression profiles (n ¼ 12 625 genes in one experiment, they allow for a ‘genome-wide’ probe sets) in diagnostic acute lymphoblastic leukemic cells assessment of genes that may predispose to leukemogenesis. from 228 children treated on protocols that included leukemo- DNA microarray analysis of gene expression has been used to genic agents such as etoposide, 13 of whom developed t-ML. identify distinct expression profiles that are characteristic of Expression of 68 probes, corresponding to 63 genes, was different leukemia subtypes.13,14 Studies using this method have significantly related to risk of t-ML. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Differences for Ectopic Versus Eutopic Cells
556 RBMO VOLUME 39 ISSUE 4 2019 ARTICLE Chemosensitivity and chemoresistance in endometriosis – differences for ectopic versus eutopic cells BIOGRAPHY Andres Salumets is Professor of Reproductive Medicine at the University of Tartu, and Scientific Head at the Competence Centre on Health Technologies, Tartu, Estonia. He has been involved in assisted reproduction for 20 years, first as an embryologist and later as a researcher. His major interests are endometriosis, endometrial biology and implantation. Darja Lavogina1,2,*, Külli Samuel1, Arina Lavrits1,3, Alvin Meltsov1, Deniss Sõritsa1,4,5, Ülle Kadastik6, Maire Peters1,4, Ago Rinken2, Andres Salumets1,4,7, 8 KEY MESSAGE Akt/PKB inhibitor GSK690693, CK2 inhibitor ARC-775, MAPK pathway inhibitor sorafenib, proteasome inhibitor bortezomib, and microtubule-depolymerizing toxin MMAE showed higher cytotoxicity in eutopic cells. In contrast, 10 µmol/l of the anthracycline toxin doxorubicin caused cellular death in ectopic cells more effectively than in eutopic cells, underlining the potential of doxorubicin in endometriosis research. ABSTRACT Research question: Endometriosis is a common gynaecological disease defined by the presence of endometrium-like tissue outside the uterus. This complex disease, often accompanied by severe pain and infertility, causes a significant medical and socioeconomic burden; hence, novel strategies are being sought for the treatment of endometriosis. Here, we set out to explore the cytotoxic effects of a panel of compounds to find toxins with different efficiency in eutopic versus ectopic cells, thus highlighting alterations in the corresponding molecular pathways. Design: The effect on cellular viability of 14 compounds was established in a cohort of paired eutopic and ectopic endometrial stromal cell samples from 11 patients. -

DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Laan et al. Clinical Epigenetics (2020) 12:9 https://doi.org/10.1186/s13148-019-0803-1 RESEARCH Open Access DNA methylation changes in Down syndrome derived neural iPSCs uncover co- dysregulation of ZNF and HOX3 families of transcription factors Loora Laan1†, Joakim Klar1†, Maria Sobol1, Jan Hoeber1, Mansoureh Shahsavani2, Malin Kele2, Ambrin Fatima1, Muhammad Zakaria1, Göran Annerén1, Anna Falk2, Jens Schuster1 and Niklas Dahl1* Abstract Background: Down syndrome (DS) is characterized by neurodevelopmental abnormalities caused by partial or complete trisomy of human chromosome 21 (T21). Analysis of Down syndrome brain specimens has shown global epigenetic and transcriptional changes but their interplay during early neurogenesis remains largely unknown. We differentiated induced pluripotent stem cells (iPSCs) established from two DS patients with complete T21 and matched euploid donors into two distinct neural stages corresponding to early- and mid-gestational ages. Results: Using the Illumina Infinium 450K array, we assessed the DNA methylation pattern of known CpG regions and promoters across the genome in trisomic neural iPSC derivatives, and we identified a total of 500 stably and differentially methylated CpGs that were annotated to CpG islands of 151 genes. The genes were enriched within the DNA binding category, uncovering 37 factors of importance for transcriptional regulation and chromatin structure. In particular, we observed regional epigenetic changes of the transcription factor genes ZNF69, ZNF700 and ZNF763 as well as the HOXA3, HOXB3 and HOXD3 genes. A similar clustering of differential methylation was found in the CpG islands of the HIST1 genes suggesting effects on chromatin remodeling. Conclusions: The study shows that early established differential methylation in neural iPSC derivatives with T21 are associated with a set of genes relevant for DS brain development, providing a novel framework for further studies on epigenetic changes and transcriptional dysregulation during T21 neurogenesis. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers
Cancer TNT Ashwin Ram 12/5/2017 Background: Chromatin Writers, Readers, Erasers writer effector eg. HAT, HMT reader eg. bromodomain eraser eg. HDAC, KDM The writer HAT1: A known H4 lysine 5,12 di-acetyltransferase writer siHAT1 siControl HAT1 H4 K12Ac H4 K5Ac actin Western blot for histone H4 modifications after control and HAT1 siRNA transfections. HAT1: EGF-stimulated immunoprecipitation specific specific - - HAT1 IgG HAT1 IgG - - R α non R α non EGF: + + + - - - WB: HAT1 Immunoprecipitation / WB to measure HAT1 levels +/- Heatmap of gene expression changes of all human histone EGF acetyltransferases +/- EGF and siRNA treatments shows HAT1 expression is EGF-dependent Working model of HAT1 : The oldest “new” histone acetyltransferase EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase Surprise: HAT1 also binds (a few sites) on chromatin HAT1 ChipSeq signal sits on Hist1 locus on Chromosome 6 Read Depth HAT1 bound sites (zoom) HAT1 ChIP-seq peaks cluster at Read Depth histone H4 promoters. Hist1H2BE Hist1H4D Hist1H3D Hist1H4E Is HAT1 a transcription factor for its substrate (H4)? EGF EGFR plasma membrane HAT1 H4 H3 Rbap46/48 a nuclear membrane H4 H2A H3 H2B HAT1 S phase HAT1 is required for S-phase burst of histone H4 mRNA HAT1 Hist1H4B mRNA level 50 Rbap46 45 shCont-3 H4 40 shHAT1-A7 35 shHAT1-B6 30 25 B6 A7 - - 20 15 shHAT1 shHAT1 shControl 10 Gene actin)Expression (versus 5 HAT1 0 0 2 4 6 8 10 actin hours after release from double thymidine block G1 S G2/M HAT1 loss: Life with less histones EGF -

Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights Into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle
animals Article Integrating Single-Step GWAS and Bipartite Networks Reconstruction Provides Novel Insights into Yearling Weight and Carcass Traits in Hanwoo Beef Cattle Masoumeh Naserkheil 1 , Abolfazl Bahrami 1 , Deukhwan Lee 2,* and Hossein Mehrban 3 1 Department of Animal Science, University College of Agriculture and Natural Resources, University of Tehran, Karaj 77871-31587, Iran; [email protected] (M.N.); [email protected] (A.B.) 2 Department of Animal Life and Environment Sciences, Hankyong National University, Jungang-ro 327, Anseong-si, Gyeonggi-do 17579, Korea 3 Department of Animal Science, Shahrekord University, Shahrekord 88186-34141, Iran; [email protected] * Correspondence: [email protected]; Tel.: +82-31-670-5091 Received: 25 August 2020; Accepted: 6 October 2020; Published: 9 October 2020 Simple Summary: Hanwoo is an indigenous cattle breed in Korea and popular for meat production owing to its rapid growth and high-quality meat. Its yearling weight and carcass traits (backfat thickness, carcass weight, eye muscle area, and marbling score) are economically important for the selection of young and proven bulls. In recent decades, the advent of high throughput genotyping technologies has made it possible to perform genome-wide association studies (GWAS) for the detection of genomic regions associated with traits of economic interest in different species. In this study, we conducted a weighted single-step genome-wide association study which combines all genotypes, phenotypes and pedigree data in one step (ssGBLUP). It allows for the use of all SNPs simultaneously along with all phenotypes from genotyped and ungenotyped animals. Our results revealed 33 relevant genomic regions related to the traits of interest.