DNA Methylation Changes in Down Syndrome Derived Neural Ipscs Uncover Co-Dysregulation of ZNF and HOX3 Families of Transcription
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

University of California, San Diego
UNIVERSITY OF CALIFORNIA, SAN DIEGO The post-terminal differentiation fate of RNAs revealed by next-generation sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Biomedical Sciences by Gloria Kuo Lefkowitz Committee in Charge: Professor Benjamin D. Yu, Chair Professor Richard Gallo Professor Bruce A. Hamilton Professor Miles F. Wilkinson Professor Eugene Yeo 2012 Copyright Gloria Kuo Lefkowitz, 2012 All rights reserved. The Dissertation of Gloria Kuo Lefkowitz is approved, and it is acceptable in quality and form for publication on microfilm and electronically: __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ __________________________________________________________________ Chair University of California, San Diego 2012 iii DEDICATION Ma and Ba, for your early indulgence and support. Matt and James, for choosing more practical callings. Roy, my love, for patiently sharing the ups and downs of this journey. iv EPIGRAPH It is foolish to tear one's hair in grief, as though sorrow would be made less by baldness. ~Cicero v TABLE OF CONTENTS Signature Page .............................................................................................................. iii Dedication .................................................................................................................... -

A Cell Line P53 Mutation Type UM
A Cell line p53 mutation Type UM-SCC 1 wt UM-SCC5 Exon 5, 157 GTC --> TTC Missense mutation by transversion (Valine --> Phenylalanine UM-SCC6 wt UM-SCC9 wt UM-SCC11A wt UM-SCC11B Exon 7, 242 TGC --> TCC Missense mutation by transversion (Cysteine --> Serine) UM-SCC22A Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC22B Exon 6, 220 TAT --> TGT Missense mutation by transition (Tyrosine --> Cysteine) UM-SCC38 Exon 5, 132 AAG --> AAT Missense mutation by transversion (Lysine --> Asparagine) UM-SCC46 Exon 8, 278 CCT --> CGT Missense mutation by transversion (Proline --> Alanine) B 1 Supplementary Methods Cell Lines and Cell Culture A panel of ten established HNSCC cell lines from the University of Michigan series (UM-SCC) was obtained from Dr. T. E. Carey at the University of Michigan, Ann Arbor, MI. The UM-SCC cell lines were derived from eight patients with SCC of the upper aerodigestive tract (supplemental Table 1). Patient age at tumor diagnosis ranged from 37 to 72 years. The cell lines selected were obtained from patients with stage I-IV tumors, distributed among oral, pharyngeal and laryngeal sites. All the patients had aggressive disease, with early recurrence and death within two years of therapy. Cell lines established from single isolates of a patient specimen are designated by a numeric designation, and where isolates from two time points or anatomical sites were obtained, the designation includes an alphabetical suffix (i.e., "A" or "B"). The cell lines were maintained in Eagle's minimal essential media supplemented with 10% fetal bovine serum and penicillin/streptomycin. -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Differences for Ectopic Versus Eutopic Cells
556 RBMO VOLUME 39 ISSUE 4 2019 ARTICLE Chemosensitivity and chemoresistance in endometriosis – differences for ectopic versus eutopic cells BIOGRAPHY Andres Salumets is Professor of Reproductive Medicine at the University of Tartu, and Scientific Head at the Competence Centre on Health Technologies, Tartu, Estonia. He has been involved in assisted reproduction for 20 years, first as an embryologist and later as a researcher. His major interests are endometriosis, endometrial biology and implantation. Darja Lavogina1,2,*, Külli Samuel1, Arina Lavrits1,3, Alvin Meltsov1, Deniss Sõritsa1,4,5, Ülle Kadastik6, Maire Peters1,4, Ago Rinken2, Andres Salumets1,4,7, 8 KEY MESSAGE Akt/PKB inhibitor GSK690693, CK2 inhibitor ARC-775, MAPK pathway inhibitor sorafenib, proteasome inhibitor bortezomib, and microtubule-depolymerizing toxin MMAE showed higher cytotoxicity in eutopic cells. In contrast, 10 µmol/l of the anthracycline toxin doxorubicin caused cellular death in ectopic cells more effectively than in eutopic cells, underlining the potential of doxorubicin in endometriosis research. ABSTRACT Research question: Endometriosis is a common gynaecological disease defined by the presence of endometrium-like tissue outside the uterus. This complex disease, often accompanied by severe pain and infertility, causes a significant medical and socioeconomic burden; hence, novel strategies are being sought for the treatment of endometriosis. Here, we set out to explore the cytotoxic effects of a panel of compounds to find toxins with different efficiency in eutopic versus ectopic cells, thus highlighting alterations in the corresponding molecular pathways. Design: The effect on cellular viability of 14 compounds was established in a cohort of paired eutopic and ectopic endometrial stromal cell samples from 11 patients. -

Download Download
Supplementary Figure S1. Results of flow cytometry analysis, performed to estimate CD34 positivity, after immunomagnetic separation in two different experiments. As monoclonal antibody for labeling the sample, the fluorescein isothiocyanate (FITC)- conjugated mouse anti-human CD34 MoAb (Mylteni) was used. Briefly, cell samples were incubated in the presence of the indicated MoAbs, at the proper dilution, in PBS containing 5% FCS and 1% Fc receptor (FcR) blocking reagent (Miltenyi) for 30 min at 4 C. Cells were then washed twice, resuspended with PBS and analyzed by a Coulter Epics XL (Coulter Electronics Inc., Hialeah, FL, USA) flow cytometer. only use Non-commercial 1 Supplementary Table S1. Complete list of the datasets used in this study and their sources. GEO Total samples Geo selected GEO accession of used Platform Reference series in series samples samples GSM142565 GSM142566 GSM142567 GSM142568 GSE6146 HG-U133A 14 8 - GSM142569 GSM142571 GSM142572 GSM142574 GSM51391 GSM51392 GSE2666 HG-U133A 36 4 1 GSM51393 GSM51394 only GSM321583 GSE12803 HG-U133A 20 3 GSM321584 2 GSM321585 use Promyelocytes_1 Promyelocytes_2 Promyelocytes_3 Promyelocytes_4 HG-U133A 8 8 3 GSE64282 Promyelocytes_5 Promyelocytes_6 Promyelocytes_7 Promyelocytes_8 Non-commercial 2 Supplementary Table S2. Chromosomal regions up-regulated in CD34+ samples as identified by the LAP procedure with the two-class statistics coded in the PREDA R package and an FDR threshold of 0.5. Functional enrichment analysis has been performed using DAVID (http://david.abcc.ncifcrf.gov/) -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Anti-Histone H2B Antibody (ARG41373)
Product datasheet [email protected] ARG41373 Package: 100 μl anti-Histone H2B antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes Histone H2B Tested Reactivity Hu, Ms, Rat Tested Application FACS, ICC/IF, IHC-P, IP, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name Histone H2B Antigen Species Human Immunogen Synthetic peptide derived from Human Histone H2B. Conjugation Un-conjugated Alternate Names Histone H2B type 1-K; H2BFAiii; H2BFT; H2B/S; H2B K; H2BK; HIRA-interacting protein 1 Application Instructions Application table Application Dilution FACS 1:50 ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 IP 1:30 WB 1:5000 - 1:20000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 14 kDa Observed Size ~ 14 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.4), 150 mM NaCl, 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot www.arigobio.com 1/2 and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol HIST1H2BK Gene Full Name histone cluster 1, H2bk Background Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. -

Histone-Related Genes Are Hypermethylated in Lung Cancer
Published OnlineFirst October 1, 2019; DOI: 10.1158/0008-5472.CAN-19-1019 Cancer Genome and Epigenome Research Histone-Related Genes Are Hypermethylated in Lung Cancer and Hypermethylated HIST1H4F Could Serve as a Pan-Cancer Biomarker Shihua Dong1,Wei Li1, Lin Wang2, Jie Hu3,Yuanlin Song3, Baolong Zhang1, Xiaoguang Ren1, Shimeng Ji3, Jin Li1, Peng Xu1, Ying Liang1, Gang Chen4, Jia-Tao Lou2, and Wenqiang Yu1 Abstract Lung cancer is the leading cause of cancer-related deaths lated in all 17 tumor types from TCGA datasets (n ¼ 7,344), worldwide. Cytologic examination is the current "gold stan- which was further validated in nine different types of cancer dard" for lung cancer diagnosis, however, this has low sensi- (n ¼ 243). These results demonstrate that HIST1H4F can tivity. Here, we identified a typical methylation signature of function as a universal-cancer-only methylation (UCOM) histone genes in lung cancer by whole-genome DNA methyl- marker, which may aid in understanding general tumorigen- ation analysis, which was validated by The Cancer Genome esis and improve screening for early cancer diagnosis. Atlas (TCGA) lung cancer cohort (n ¼ 907) and was further confirmed in 265 bronchoalveolar lavage fluid samples with Significance: These findings identify a new biomarker for specificity and sensitivity of 96.7% and 87.0%, respectively. cancer detection and show that hypermethylation of histone- More importantly, HIST1H4F was universally hypermethy- related genes seems to persist across cancers. Introduction to its low specificity, LDCT is far from satisfactory as a screening tool for clinical application, similar to other currently used cancer Lung cancer is one of the most common malignant tumors and biomarkers, such as carcinoembryonic antigen (CEA), neuron- the leading cause of cancer-related deaths worldwide (1, 2). -

Brain Region-Dependent Gene Networks Associated with Selective Breeding for Increased Voluntary Wheel-Running Behavior
UC Riverside UC Riverside Previously Published Works Title Brain region-dependent gene networks associated with selective breeding for increased voluntary wheel-running behavior. Permalink https://escholarship.org/uc/item/8c49g8fd Journal PloS one, 13(8) ISSN 1932-6203 Authors Zhang, Pan Rhodes, Justin S Garland, Theodore et al. Publication Date 2018 DOI 10.1371/journal.pone.0201773 Peer reviewed eScholarship.org Powered by the California Digital Library University of California RESEARCH ARTICLE Brain region-dependent gene networks associated with selective breeding for increased voluntary wheel-running behavior Pan Zhang1,2, Justin S. Rhodes3,4, Theodore Garland, Jr.5, Sam D. Perez3, Bruce R. Southey2, Sandra L. Rodriguez-Zas2,6,7* 1 Illinois Informatics Institute, University of Illinois at Urbana-Champaign, Urbana, IL, United States of America, 2 Department of Animal Sciences, University of Illinois at Urbana-Champaign, Urbana, IL, United a1111111111 States of America, 3 Beckman Institute for Advanced Science and Technology, Urbana, IL, United States of a1111111111 America, 4 Center for Nutrition, Learning and Memory, University of Illinois at Urbana-Champaign, Urbana, a1111111111 IL, United States of America, 5 Department of Evolution, Ecology, and Organismal Biology, University of a1111111111 California, Riverside, CA, United States of America, 6 Department of Statistics, University of Illinois at Urbana-Champaign, Urbana, IL, United States of America, 7 Carle Woese Institute for Genomic Biology, a1111111111 University of Illinois at Urbana-Champaign, Urbana, IL, United States of America * [email protected] OPEN ACCESS Abstract Citation: Zhang P, Rhodes JS, Garland T, Jr., Perez SD, Southey BR, Rodriguez-Zas SL (2018) Brain Mouse lines selectively bred for high voluntary wheel-running behavior are helpful models region-dependent gene networks associated with for uncovering gene networks associated with increased motivation for physical activity and selective breeding for increased voluntary wheel- other reward-dependent behaviors. -

A Yeast Phenomic Model for the Influence of Warburg Metabolism on Genetic Buffering of Doxorubicin Sean M
Santos and Hartman Cancer & Metabolism (2019) 7:9 https://doi.org/10.1186/s40170-019-0201-3 RESEARCH Open Access A yeast phenomic model for the influence of Warburg metabolism on genetic buffering of doxorubicin Sean M. Santos and John L. Hartman IV* Abstract Background: The influence of the Warburg phenomenon on chemotherapy response is unknown. Saccharomyces cerevisiae mimics the Warburg effect, repressing respiration in the presence of adequate glucose. Yeast phenomic experiments were conducted to assess potential influences of Warburg metabolism on gene-drug interaction underlying the cellular response to doxorubicin. Homologous genes from yeast phenomic and cancer pharmacogenomics data were analyzed to infer evolutionary conservation of gene-drug interaction and predict therapeutic relevance. Methods: Cell proliferation phenotypes (CPPs) of the yeast gene knockout/knockdown library were measured by quantitative high-throughput cell array phenotyping (Q-HTCP), treating with escalating doxorubicin concentrations under conditions of respiratory or glycolytic metabolism. Doxorubicin-gene interaction was quantified by departure of CPPs observed for the doxorubicin-treated mutant strain from that expected based on an interaction model. Recursive expectation-maximization clustering (REMc) and Gene Ontology (GO)-based analyses of interactions identified functional biological modules that differentially buffer or promote doxorubicin cytotoxicity with respect to Warburg metabolism. Yeast phenomic and cancer pharmacogenomics data were integrated to predict differential gene expression causally influencing doxorubicin anti-tumor efficacy. Results: Yeast compromised for genes functioning in chromatin organization, and several other cellular processes are more resistant to doxorubicin under glycolytic conditions. Thus, the Warburg transition appears to alleviate requirements for cellular functions that buffer doxorubicin cytotoxicity in a respiratory context. -
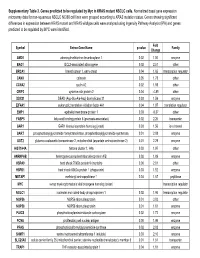
Supplementary Table 3. Genes Predicted to Be Regulated by Myc in KRAS Mutant NSCLC Cells
Supplementary Table 3. Genes predicted to be regulated by Myc in KRAS mutant NSCLC cells. Normalized basal gene expression microarray data for non-squamous NSCLC NCI60 cell lines were grouped according to KRAS mutation status. Genes showing significant differences in expression between KRAS mutant and KRAS wildtype cells were analyzed using Ingenuity Pathway Analysis (IPA) and genes predicted to be regulated by MYC were identified. Fold Symbol Entrez Gene Name p-value Family Change AMD1 adenosylmethionine decarboxylase 1 0.02 1.50 enzyme BAG1 BCL2-associated athanogene 0.02 2.57 other BRCA1 breast cancer 1, early onset 0.04 1.65 transcription regulator CANX calnexin 0.05 1.78 other CCNA2 cyclin A2 0.02 1.98 other CRIP2 cysteine-rich protein 2 0.04 -4.89 other DDX21 DEAD (Asp-Glu-Ala-Asp) box helicase 21 0.03 1.59 enzyme EIF4A1 eukaryotic translation initiation factor 4A1 0.04 1.87 translation regulator EMP1 epithelial membrane protein 1 0.03 -6.37 other FABP5 fatty acid binding protein 5 (psoriasis-associated) 0.02 2.26 transporter GAR1 GAR1 ribonucleoprotein homolog (yeast) 0.03 1.56 ion channel GART phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, 0.01 2.08 enzyme GOT2 glutamic-oxaloacetic transaminase 2, mitochondrial (aspartate aminotransferase 2) 0.01 2.29 enzyme HIST1H4A histone cluster 1, H4a 0.03 1.97 other HNRNPAB heterogeneous nuclear ribonucleoprotein A/B 0.02 1.89 enzyme HSPA9 heat shock 70kDa protein 9 (mortalin) 0.00 2.01 other HSPD1 heat shock 60kDa protein 1 (chaperonin) 0.03 1.52 enzyme -

A Mutation in Histone H2B Represents a New Class of Oncogenic Driver
Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. A Mutation in Histone H2B Represents A New Class Of Oncogenic Driver Richard L. Bennett1, Aditya Bele1, Eliza C. Small2, Christine M. Will2, Behnam Nabet3, Jon A. Oyer2, Xiaoxiao Huang1,9, Rajarshi P. Ghosh4, Adrian T. Grzybowski5, Tao Yu6, Qiao Zhang7, Alberto Riva8, Tanmay P. Lele7, George C. Schatz9, Neil L. Kelleher9 Alexander J. Ruthenburg5, Jan Liphardt4 and Jonathan D. Licht1 * 1 Division of Hematology/Oncology, University of Florida Health Cancer Center, Gainesville, FL 2 Division of Hematology/Oncology, Northwestern University 3 Department of Cancer Biology, Dana Farber Cancer Institute and Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School 4 Department of Bioengineering, Stanford University 5 Department of Molecular Genetics and Cell Biology, The University of Chicago 6 Department of Chemistry, Tennessee Technological University 7 Department of Chemical Engineering, University of Florida 8 Bioinformatics Core, Interdisciplinary Center for Biotechnology Research, University of Florida 9 Department of Chemistry, Northwestern University, Evanston IL 60208 Running title: Histone mutations in cancer *Corresponding Author: Jonathan D. Licht, MD The University of Florida Health Cancer Center Cancer and Genetics Research Complex, Suite 145 2033 Mowry Road Gainesville, FL 32610 352-273-8143 [email protected] Disclosures: The authors have no conflicts of interest to declare Downloaded from cancerdiscovery.aacrjournals.org on September 27, 2021. © 2019 American Association for Cancer Research. Author Manuscript Published OnlineFirst on July 23, 2019; DOI: 10.1158/2159-8290.CD-19-0393 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited.