New Insights on Human Essential Genes Based on Integrated Multi
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Polyphosphate-Dependent Synthesis of ATP and ADP by the Family-2 Polyphosphate Kinases in Bacteria
Polyphosphate-dependent synthesis of ATP and ADP by the family-2 polyphosphate kinases in bacteria Boguslaw Noceka, Samvel Kochinyanb, Michael Proudfootb, Greg Brownb, Elena Evdokimovaa,b, Jerzy Osipiuka, Aled M. Edwardsa,b, Alexei Savchenkoa,b, Andrzej Joachimiaka,c,1, and Alexander F. Yakuninb,1 aMidwest Center for Structural Genomics and Structural Biology Center, Department of Biosciences, Argonne National Laboratory, 9700 South Cass Avenue, Building 202, Argonne, IL 60439; bBanting and Best Department of Medical Research, University of Toronto, Toronto, ON, Canada M5G 1L6; and cDepartment of Biochemistry and Molecular Biology, University of Chicago, 920 East 58th Street, Chicago, IL 60637 Edited by Robert Haselkorn, University of Chicago, Chicago, IL, and approved September 30, 2008 (received for review August 1, 2008) Inorganic polyphosphate (polyP) is a linear polymer of tens or hun- exopolysaccharide essential for the virulence of P. aeruginosa (12). dreds of phosphate residues linked by high-energy bonds. It is found In the social slime mold Dictyostelium discoideum, a different PPK in all organisms and has been proposed to serve as an energy source was found (DdPPK2) with sequence and properties similar to that in a pre-ATP world. This ubiquitous and abundant biopolymer plays of actin-related proteins (Arps) (14). DdPPK2 is a complex of 3 numerous and vital roles in metabolism and regulation in prokaryotes Arps (Arp1, Arp2, and Arpx) and resembles the actin family in and eukaryotes, but the underlying molecular mechanisms for most molecular weight, sequence, and filamentous structure. Remark- activities of polyP remain unknown. In prokaryotes, the synthesis and ably, DdPPK2 can polymerize into an actin-like filament concurrent utilization of polyP are catalyzed by 2 families of polyP kinases, PPK1 with the synthesis of polyP (14). -

Regulation and Dysregulation of Chromosome Structure in Cancer
Regulation and Dysregulation of Chromosome Structure in Cancer The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Hnisz, Denes et al. “Regulation and Dysregulation of Chromosome Structure in Cancer.” Annual Review of Cancer Biology 2, 1 (March 2018): 21–40 © 2018 Annual Reviews As Published https://doi.org/10.1146/annurev-cancerbio-030617-050134 Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/117286 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike Detailed Terms http://creativecommons.org/licenses/by-nc-sa/4.0/ Regulation and dysregulation of chromosome structure in cancer Denes Hnisz1*, Jurian Schuijers1, Charles H. Li1,2, Richard A. Young1,2* 1 Whitehead Institute for Biomedical Research, 455 Main Street, Cambridge, MA 02142, USA 2 Department of Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA * Corresponding authors Corresponding Authors: Denes Hnisz Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-7181 Fax: (617) 258-0376 [email protected] Richard A. Young Whitehead Institute for Biomedical Research 455 Main Street Cambridge, MA 02142 Tel: (617) 258-5218 Fax: (617) 258-0376 [email protected] 1 Summary Cancer arises from genetic alterations that produce dysregulated gene expression programs. Normal gene regulation occurs in the context of chromosome loop structures called insulated neighborhoods, and recent studies have shown that these structures are altered and can contribute to oncogene dysregulation in various cancer cells. We review here the types of genetic and epigenetic alterations that influence neighborhood structures and contribute to gene dysregulation in cancer, present models for insulated neighborhoods associated with the most prominent human oncogenes, and discuss how such models may lead to further advances in cancer diagnosis and therapy. -

CCT3 Rabbit Pab
Leader in Biomolecular Solutions for Life Science CCT3 Rabbit pAb Catalog No.: A6547 3 Publications Basic Information Background Catalog No. The protein encoded by this gene is a molecular chaperone that is a member of the A6547 chaperonin containing TCP1 complex (CCT), also known as the TCP1 ring complex (TRiC). This complex consists of two identical stacked rings, each containing eight different Observed MW proteins. Unfolded polypeptides enter the central cavity of the complex and are folded 60kDa in an ATP-dependent manner. The complex folds various proteins, including actin and tubulin. Alternate transcriptional splice variants have been characterized for this gene. Calculated MW In addition, a pseudogene of this gene has been found on chromosome 8. 56kDa/60kDa Category Primary antibody Applications WB,IHC,IF Cross-Reactivity Human, Mouse, Rat Recommended Dilutions Immunogen Information WB 1:500 - 1:2000 Gene ID Swiss Prot 7203 P49368 IHC 1:50 - 1:200 Immunogen 1:50 - 1:200 IF Recombinant fusion protein containing a sequence corresponding to amino acids 1-300 of human CCT3 (NP_005989.3). Synonyms CCT3;CCT-gamma;CCTG;PIG48;TCP-1-gamma;TRIC5 Contact Product Information www.abclonal.com Source Isotype Purification Rabbit IgG Affinity purification Storage Store at -20℃. Avoid freeze / thaw cycles. Buffer: PBS with 0.02% sodium azide,50% glycerol,pH7.3. Validation Data Western blot analysis of extracts of various cell lines, using CCT3 antibody (A6547) at 1:1000 dilution. Secondary antibody: HRP Goat Anti-Rabbit IgG (H+L) (AS014) at 1:10000 dilution. Lysates/proteins: 25ug per lane. Blocking buffer: 3% nonfat dry milk in TBST. -

Apc11 (ANAPC11) (NM 001002245) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC223841L4 Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone Tag: mGFP Symbol: ANAPC11 Synonyms: APC11; Apc11p; HSPC214 Vector: pLenti-C-mGFP-P2A-Puro (PS100093) E. coli Selection: Chloramphenicol (34 ug/mL) Cell Selection: Puromycin ORF Nucleotide The ORF insert of this clone is exactly the same as(RC223841). Sequence: Restriction Sites: SgfI-MluI Cloning Scheme: ACCN: NM_001002245 ORF Size: 252 bp This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Apc11 (ANAPC11) (NM_001002245) Human Tagged ORF Clone – RC223841L4 OTI Disclaimer: Due to the inherent nature of this plasmid, standard methods to replicate additional amounts of DNA in E. coli are highly likely to result in mutations and/or rearrangements. Therefore, OriGene does not guarantee the capability to replicate this plasmid DNA. Additional amounts of DNA can be purchased from OriGene with batch-specific, full-sequence verification at a reduced cost. Please contact our customer care team at [email protected] or by calling 301.340.3188 option 3 for pricing and delivery. The molecular sequence of this clone aligns with the gene accession number as a point of reference only. However, individual transcript sequences of the same gene can differ through naturally occurring variations (e.g. -

View the Poster.Pdf
Identifying Novel Associations for Iron-Related Genes in High-Grade Ovarian Cancer Abigail Descoteaux*1, José W. Velázquez*2, Anna Konstorum3, Reinhard Laubenbacher3,4 1Vassar College, 2University of Puerto Rico at Cayey, 3Center for Quantitative Medicine, UConn Health, 4The Jackson Laboratory for Genomic Medicine *These authors contributed equally to this work Introduction Methods Testable Hypothesis § Iron can gain and lose electrons, making it enzymatically Microarray gene expression Adjacency matrix Community detection in a weighted, KEGG pathways Tumor cells promote an inflammatory microenvironment via [1] data from HGSOC clinical • Top 10,000 most variable genes undirected network • Manually curated biological pathways useful in cell replication, metabolism, and growth • Bicor derived from Pearson but • ORA: ask whether any known pathways • Order Statistics Local Optimization Method (OSLOM) recruitment of tumor-associated macrophages, that in turn samples less sensitive to outliers are significantly over-represented in the • Assembled unsupervised with respect to biology by locally § Cancer cells sequester iron by altering the expression of • The Cancer Genome Atlas (TCGA) • Correlation cut-off at 0.45 genes within the community secrete IL4 and CCL4 to promote an iron-influx phenotype in optimizing the statistical significance of clusters and using [1] • Affymetrix HGU133A (n=488) (FDR corrected, α=0.01) Pathway 2 several regulators of iron metabolism (Figure 1) • Tothill et al. [2] this as a fitness score ovarian cancer tumor cells. • • HGU133plus2 (n=217) Accounts for edge weight, overlapping communities, and Pathway 1 hierarchies a. Normal epithelial cell Fe3+ Fe3+ Pathway 3 Tumor-associated 3+ 3+ Tumor cells Fe Fe macrophages Hepcidin (HAMP) samples genes Biweight midcorrelation Over-representation (bicor) Hypothesis CSF1R Ferroportin OSLOM analysis (ORA) CCR1[8] (SLC40A1) generation and [7] genes genes 1. -

Gene Knockdown of CENPA Reduces Sphere Forming Ability and Stemness of Glioblastoma Initiating Cells
Neuroepigenetics 7 (2016) 6–18 Contents lists available at ScienceDirect Neuroepigenetics journal homepage: www.elsevier.com/locate/nepig Gene knockdown of CENPA reduces sphere forming ability and stemness of glioblastoma initiating cells Jinan Behnan a,1, Zanina Grieg b,c,1, Mrinal Joel b,c, Ingunn Ramsness c, Biljana Stangeland a,b,⁎ a Department of Molecular Medicine, Institute of Basic Medical Sciences, The Medical Faculty, University of Oslo, Oslo, Norway b Norwegian Center for Stem Cell Research, Department of Immunology and Transfusion Medicine, Oslo University Hospital, Oslo, Norway c Vilhelm Magnus Laboratory for Neurosurgical Research, Institute for Surgical Research and Department of Neurosurgery, Oslo University Hospital, Oslo, Norway article info abstract Article history: CENPA is a centromere-associated variant of histone H3 implicated in numerous malignancies. However, the Received 20 May 2016 role of this protein in glioblastoma (GBM) has not been demonstrated. GBM is one of the most aggressive Received in revised form 23 July 2016 human cancers. GBM initiating cells (GICs), contained within these tumors are deemed to convey Accepted 2 August 2016 characteristics such as invasiveness and resistance to therapy. Therefore, there is a strong rationale for targeting these cells. We investigated the expression of CENPA and other centromeric proteins (CENPs) in Keywords: fi CENPA GICs, GBM and variety of other cell types and tissues. Bioinformatics analysis identi ed the gene signature: fi Centromeric proteins high_CENP(AEFNM)/low_CENP(BCTQ) whose expression correlated with signi cantly worse GBM patient Glioblastoma survival. GBM Knockdown of CENPA reduced sphere forming ability, proliferation and cell viability of GICs. We also Brain tumor detected significant reduction in the expression of stemness marker SOX2 and the proliferation marker Glioblastoma initiating cells and therapeutic Ki67. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

) (51) International Patent Classification: Columbia V5G 1G3
) ( (51) International Patent Classification: Columbia V5G 1G3 (CA). PANDEY, Nihar R.; 10209 A 61K 31/4525 (2006.01) C07C 39/23 (2006.01) 128A St, Surrey, British Columbia V3T 3E7 (CA). A61K 31/05 (2006.01) C07D 405/06 (2006.01) (74) Agent: ZIESCHE, Sonia et al.; Gowling WLG (Canada) A61P25/22 (2006.01) LLP, 2300 - 550 Burrard Street, Vancouver, British Colum¬ (21) International Application Number: bia V6C 2B5 (CA). PCT/CA2020/050165 (81) Designated States (unless otherwise indicated, for every (22) International Filing Date: kind of national protection av ailable) . AE, AG, AL, AM, 07 February 2020 (07.02.2020) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (25) Filing Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, (26) Publication Language: English HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (30) Priority Data: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 16/270,389 07 February 2019 (07.02.2019) US OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (63) Related by continuation (CON) or continuation-in-part SC, SD, SE, SG, SK, SL, ST, SV, SY, TH, TJ, TM, TN, TR, (CIP) to earlier application: TT, TZ, UA, UG, US, UZ, VC, VN, WS, ZA, ZM, ZW. US 16/270,389 (CON) (84) Designated States (unless otherwise indicated, for every Filed on 07 Februaiy 2019 (07.02.2019) kind of regional protection available) . -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -
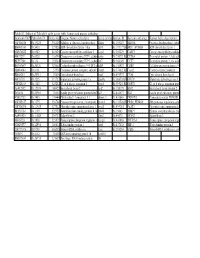
Table S3: Subset of Zebrafish Early Genes with Human And
Table S3: Subset of Zebrafish early genes with human and mouse orthologs Genbank ID(ZFZebrafish ID Entrez GenUnigene Name (zebrafish) Gene symbo Human ID Humann ortholog Human Gene description AW116838 Dr.19225 336425 Aldolase a, fructose-bisphosphate aldoa Hs.155247 ALDOA Fructose-bisphosphate aldola BM005100 Dr.5438 327026 ADP-ribosylation factor 1 like arf1l Hs.119177||HsARF1_HUMAN ADP-ribosylation factor 1 AW076882 Dr.6582 403025 Cancer susceptibility candidate 3 casc3 Hs.350229 CASC3 Cancer susceptibility candidat AI437239 Dr.6928 116994 Chaperonin containing TCP1, subun cct6a Hs.73072||Hs.CCT6A T-complex protein 1, zeta sub BE557308 Dr.134 192324 Chaperonin containing TCP1, subun cct7 Hs.368149 CCT7 T-complex protein 1, eta subu BG303647 Dr.26326 321602 Cyclin-dependent kinase 9 (CDC2-recdk9 Hs.150423 CDK9 Cell division protein kinase 9 AB040044 Dr.8161 57970 Coatomer protein complex, subunit zcopz1 Hs.37482||Hs.Copz2 Coatomer zeta-2 subunit BI888253 Dr.20911 30436 Eyes absent homolog 1 eya1 Hs.491997 EYA4 Eyes absent homolog 4 AI878758 Dr.3225 317737 Glutamate dehydrogenase 1a glud1a Hs.368538||HsGLUD1 Glutamate dehydrogenase 1, AW128619 Dr.1388 325284 G1 to S phase transition 1 gspt1 Hs.59523||Hs.GSPT1 G1 to S phase transition prote AF412832 Dr.12595 140427 Heat shock factor 2 hsf2 Hs.158195 HSF2 Heat shock factor protein 2 D38454 Dr.20916 30151 Insulin gene enhancer protein Islet3 isl3 Hs.444677 ISL2 Insulin gene enhancer protein AY052752 Dr.7485 170444 Pbx/knotted 1 homeobox 1.1 pknox1.1 Hs.431043 PKNOX1 Homeobox protein PKNOX1 -

Genomic Amplification of Chromosome 20Q13.33 Is the Early Biomarker For
Bui et al. BMC Medical Genomics 2020, 13(Suppl 10):149 https://doi.org/10.1186/s12920-020-00776-z RESEARCH Open Access Genomic amplification of chromosome 20q13.33 is the early biomarker for the development of sporadic colorectal carcinoma Vo-Minh-Hoang Bui1,2, Clément Mettling3, Jonathan Jou4 and H. Sunny Sun1,5* From The 18th Asia Pacific Bioinformatics Conference Seoul, Korea. 18-20 August 2020 Abstract Background: Colorectal carcinoma (CRC) is the third most common cancer in the world and also the third leading cause of cancer-related mortality in Taiwan. CRC tumorigenesis is a multistep process, starting from mutations causing loss of function of tumor suppressor genes, canonically demonstrated in adenomatous polyposis coli pathogenesis. Although many genes or chromosomal alterations have been shown to be involved in this process, there are still unrecognized molecular events within CRC tumorigenesis. Elucidating these mechanisms may help improve the management and treatment. Methods: In this study, we aimed to identify copy number alteration of the smallest chromosomal regions that is significantly associated with sporadic CRC tumorigenesis using high-resolution array-based Comparative Genomic Hybridization (aCGH) and quantitative Polymerase chain reaction (qPCR). In addition, microsatellite instability assay and sequencing-based mutation assay were performed to illustrate the initiation event of CRC tumorigenesis. Results: A total of 571 CRC patients were recruited and 377 paired CRC tissues from sporadic CRC cases were used to define the smallest regions with chromosome copy number changes. In addition, 198 colorectal polyps from 160 patients were also used to study the role of 20q13.33 gain in CRC tumorigenesis. -

IL21R Expressing CD14+CD16+ Monocytes Expand in Multiple
Plasma Cell Disorders SUPPLEMENTARY APPENDIX IL21R expressing CD14 +CD16 + monocytes expand in multiple myeloma patients leading to increased osteoclasts Marina Bolzoni, 1 Domenica Ronchetti, 2,3 Paola Storti, 1,4 Gaetano Donofrio, 5 Valentina Marchica, 1,4 Federica Costa, 1 Luca Agnelli, 2,3 Denise Toscani, 1 Rosanna Vescovini, 1 Katia Todoerti, 6 Sabrina Bonomini, 7 Gabriella Sammarelli, 1,7 Andrea Vecchi, 8 Daniela Guasco, 1 Fabrizio Accardi, 1,7 Benedetta Dalla Palma, 1,7 Barbara Gamberi, 9 Carlo Ferrari, 8 Antonino Neri, 2,3 Franco Aversa 1,4,7 and Nicola Giuliani 1,4,7 1Myeloma Unit, Dept. of Medicine and Surgery, University of Parma; 2Dept. of Oncology and Hemato-Oncology, University of Milan; 3Hematology Unit, “Fondazione IRCCS Ca’ Granda”, Ospedale Maggiore Policlinico, Milan; 4CoreLab, University Hospital of Parma; 5Dept. of Medical-Veterinary Science, University of Parma; 6Laboratory of Pre-clinical and Translational Research, IRCCS-CROB, Referral Cancer Center of Basilicata, Rionero in Vulture; 7Hematology and BMT Center, University Hospital of Parma; 8Infectious Disease Unit, University Hospital of Parma and 9“Dip. Oncologico e Tecnologie Avanzate”, IRCCS Arcispedale Santa Maria Nuova, Reggio Emilia, Italy ©2017 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.153841 Received: August 5, 2016. Accepted: December 23, 2016. Pre-published: January 5, 2017. Correspondence: [email protected] SUPPLEMENTAL METHODS Immunophenotype of BM CD14+ in patients with monoclonal gammopathies. Briefly, 100 μl of total BM aspirate was incubated in the dark with anti-human HLA-DR-PE (clone L243; BD), anti-human CD14-PerCP-Cy 5.5, anti-human CD16-PE-Cy7 (clone B73.1; BD) and anti-human CD45-APC-H 7 (clone 2D1; BD) for 20 min.