Standard Thermodynamic Values
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

Energy and Enthalpy Thermodynamics
Energy and Energy and Enthalpy Thermodynamics The internal energy (E) of a system consists of The energy change of a reaction the kinetic energy of all the particles (related to is measured at constant temperature) plus the potential energy of volume (in a bomb interaction between the particles and within the calorimeter). particles (eg bonding). We can only measure the change in energy of the system (units = J or Nm). More conveniently reactions are performed at constant Energy pressure which measures enthalpy change, ∆H. initial state final state ∆H ~ ∆E for most reactions we study. final state initial state ∆H < 0 exothermic reaction Energy "lost" to surroundings Energy "gained" from surroundings ∆H > 0 endothermic reaction < 0 > 0 2 o Enthalpy of formation, fH Hess’s Law o Hess's Law: The heat change in any reaction is the The standard enthalpy of formation, fH , is the change in enthalpy when one mole of a substance is formed from same whether the reaction takes place in one step or its elements under a standard pressure of 1 atm. several steps, i.e. the overall energy change of a reaction is independent of the route taken. The heat of formation of any element in its standard state is defined as zero. o The standard enthalpy of reaction, H , is the sum of the enthalpy of the products minus the sum of the enthalpy of the reactants. Start End o o o H = prod nfH - react nfH 3 4 Example Application – energy foods! Calculate Ho for CH (g) + 2O (g) CO (g) + 2H O(l) Do you get more energy from the metabolism of 1.0 g of sugar or -

Chemistry 130 Gibbs Free Energy
Chemistry 130 Gibbs Free Energy Dr. John F. C. Turner 409 Buehler Hall [email protected] Chemistry 130 Equilibrium and energy So far in chemistry 130, and in Chemistry 120, we have described chemical reactions thermodynamically by using U - the change in internal energy, U, which involves heat transferring in or out of the system only or H - the change in enthalpy, H, which involves heat transfers in and out of the system as well as changes in work. U applies at constant volume, where as H applies at constant pressure. Chemistry 130 Equilibrium and energy When chemical systems change, either physically through melting, evaporation, freezing or some other physical process variables (V, P, T) or chemically by reaction variables (ni) they move to a point of equilibrium by either exothermic or endothermic processes. Characterizing the change as exothermic or endothermic does not tell us whether the change is spontaneous or not. Both endothermic and exothermic processes are seen to occur spontaneously. Chemistry 130 Equilibrium and energy Our descriptions of reactions and other chemical changes are on the basis of exothermicity or endothermicity ± whether H is negative or positive H is negative ± exothermic H is positive ± endothermic As a description of changes in heat content and work, these are adequate but they do not describe whether a process is spontaneous or not. There are endothermic processes that are spontaneous ± evaporation of water, the dissolution of ammonium chloride in water, the melting of ice and so on. We need a thermodynamic description of spontaneous processes in order to fully describe a chemical system Chemistry 130 Equilibrium and energy A spontaneous process is one that takes place without any influence external to the system. -

Lecture 13. Thermodynamic Potentials (Ch
Lecture 13. Thermodynamic Potentials (Ch. 5) So far, we have been using the total internal energy U and, sometimes, the enthalpy H to characterize various macroscopic systems. These functions are called the thermodynamic potentials: all the thermodynamic properties of the system can be found by taking partial derivatives of the TP. For each TP, a set of so-called “natural variables” exists: −= + μ NddVPSdTUd = + + μ NddPVSdTHd Today we’ll introduce the other two thermodynamic potentials: theHelmhotzfree energy F and Gibbs free energy G. Depending on the type of a process, one of these four thermodynamic potentials provides the most convenient description (and is tabulated). All four functions have units of energy. When considering different types of Potential Variables processes, we will be interested in two main U (S,V,N) S, V, N issues: H (S,P,N) S, P, N what determines the stability of a system and how the system evolves towards an F (T,V,N) V, T, N equilibrium; G (T,P,N) P, T, N how much work can be extracted from a system. Diffusive Equilibrium and Chemical Potential For completeness, let’s rcall what we’vee learned about the chemical potential. 1 P μ d U T d= S− P+ μ dVd d= S Nd U +dV d − N T T T The meaning of the partial derivative (∂S/∂N)U,V : let’s fix VA and VB (the membrane’s position is fixed), but U , V , S U , V , S A A A B B B assume that the membrane becomes permeable for gas molecules (exchange of both U and N between the sub- ns ilesystems, the molecuA and B are the same ). -

Thermodynamic Potentials and Thermodynamic Relations In
arXiv:1004.0337 Thermodynamic potentials and Thermodynamic Relations in Nonextensive Thermodynamics Guo Lina, Du Jiulin Department of Physics, School of Science, Tianjin University, Tianjin 300072, China Abstract The generalized Gibbs free energy and enthalpy is derived in the framework of nonextensive thermodynamics by using the so-called physical temperature and the physical pressure. Some thermodynamical relations are studied by considering the difference between the physical temperature and the inverse of Lagrange multiplier. The thermodynamical relation between the heat capacities at a constant volume and at a constant pressure is obtained using the generalized thermodynamical potential, which is found to be different from the traditional one in Gibbs thermodynamics. But, the expressions for the heat capacities using the generalized thermodynamical potentials are unchanged. PACS: 05.70.-a; 05.20.-y; 05.90.+m Keywords: Nonextensive thermodynamics; The generalized thermodynamical relations 1. Introduction In the traditional thermodynamics, there are several fundamental thermodynamic potentials, such as internal energy U , Helmholtz free energy F , enthalpy H , Gibbs free energyG . Each of them is a function of temperature T , pressure P and volume V . They and their thermodynamical relations constitute the basis of classical thermodynamics. Recently, nonextensive thermo-statitstics has attracted significent interests and has obtained wide applications to so many interesting fields, such as astrophysics [2, 1 3], real gases [4], plasma [5], nuclear reactions [6] and so on. Especially, one has been studying the problems whether the thermodynamic potentials and their thermo- dynamic relations in nonextensive thermodynamics are the same as those in the classical thermodynamics [7, 8]. In this paper, under the framework of nonextensive thermodynamics, we study the generalized Gibbs free energy Gq in section 2, the heat capacity at constant volume CVq and heat capacity at constant pressure CPq in section 3, and the generalized enthalpy H q in Sec.4. -

Enthalpy and Free Energy of Reaction
KINETICS AND THERMODYNAMICS OF REACTIONS Key words: Enthalpy, entropy, Gibbs free energy, heat of reaction, energy of activation, rate of reaction, rate law, energy profiles, order and molecularity, catalysts INTRODUCTION This module offers a very preliminary detail of basic thermodynamics. Emphasis is given on the commonly used terms such as free energy of a reaction, rate of a reaction, multi-step reactions, intermediates, transition states etc., FIRST LAW OF THERMODYNAMICS. o Law of conservation of energy. States that, • Energy can be neither created nor destroyed. • Total energy of the universe remains constant. • Energy can be converted from one form to another form. eg. Combustion of octane (petrol). 2 C8H18(l) + 25 O2(g) CO2(g) + 18 H2O(g) Conversion of potential energy into thermal energy. → 16 + 10.86 KJ/mol . ESSENTIAL FACTORS FOR REACTION. For a reaction to progress. • The equilibrium must favor the products- • Thermodynamics(energy difference between reactant and product) should be favorable • Reaction rate must be fast enough to notice product formation in a reasonable period. • Kinetics( rate of reaction) ESSENTIAL TERMS OF THERMODYNAMICS. Thermodynamics. Predicts whether the reaction is thermally favorable. The energy difference between the final products and reactants are taken as the guiding principle. The equilibrium will be in favor of products when the product energy is lower. Molecule with lowered energy posses enhanced stability. Essential terms o Free energy change (∆G) – Overall free energy difference between the reactant and the product o Enthalpy (∆H) – Heat content of a system under a given pressure. o Entropy (∆S) – The energy of disorderness, not available for work in a thermodynamic process of a system. -

Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions
CHEM 1B: GENERAL CHEMISTRY Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions Instructor: Dr. Orlando E. Raola 20-1 Santa Rosa Junior College Chapter 20 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20-2 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20.1 The Second Law of Thermodynamics: Predicting Spontaneous Change 20.2 Calculating Entropy Change of a Reaction 20.3 Entropy, Free Energy, and Work 20.4 Free Energy, Equilibrium, and Reaction Direction 20-3 Spontaneous Change A spontaneous change is one that occurs without a continuous input of energy from outside the system. All chemical processes require energy (activation energy) to take place, but once a spontaneous process has begun, no further input of energy is needed. A nonspontaneous change occurs only if the surroundings continuously supply energy to the system. If a change is spontaneous in one direction, it will be nonspontaneous in the reverse direction. 20-4 The First Law of Thermodynamics Does Not Predict Spontaneous Change Energy is conserved. It is neither created nor destroyed, but is transferred in the form of heat and/or work. DE = q + w The total energy of the universe is constant: DEsys = -DEsurr or DEsys + DEsurr = DEuniv = 0 The law of conservation of energy applies to all changes, and does not allow us to predict the direction of a spontaneous change. 20-5 DH Does Not Predict Spontaneous Change A spontaneous change may be exothermic or endothermic. Spontaneous exothermic processes include: • freezing and condensation at low temperatures, • combustion reactions, • oxidation of iron and other metals. -
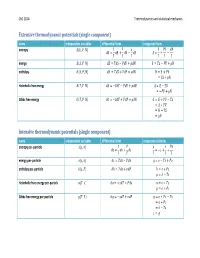
Thermodynamic Calculus Manipulations Name Applies to
ChE 210A Thermodynamics and statistical mechanics Extensive thermodynamic potentials (single component) name independent variables differential form integrated form entropy ͍ʚ̿, ͐, ͈ʛ 1 ͊ ̿ ͊͐ ͈ ͍͘ Ɣ ̿͘ ƍ ͐͘ Ǝ ͈͘ ͍ Ɣ ƍ Ǝ ͎ ͎ ͎ ͎ ͎ ͎ energy ̿ʚ͍, ͐, ͈ʛ ̿͘ Ɣ ͎͍͘ Ǝ ͊͐͘ ƍ ͈͘ ̿ Ɣ ͎͍ Ǝ ͊͐ ƍ ͈ enthalpy ͂ʚ͍, ͊, ͈ʛ ͂͘ Ɣ ͎͍͘ ƍ ͐͊͘ ƍ ͈͘ ͂ Ɣ ̿ ƍ ͊͐ Ɣ ͎͍ ƍ ͈ Helmholtz free energy ̻ʚ͎, ͐, ͈ʛ ̻͘ Ɣ Ǝ͍͎͘ Ǝ ͊͐͘ ƍ ͈͘ ̻ Ɣ ̿ Ǝ ͎͍ Ɣ Ǝ͊͐ ƍ ͈ Gibbs free energy ́ʚ͎, ͊, ͈ʛ ́͘ Ɣ Ǝ͍͎͘ ƍ ͐͊͘ ƍ ͈͘ ́ Ɣ ̿ ƍ ͊͐ Ǝ ͎͍ Ɣ ̻ ƍ ͊͐ Ɣ ͂ Ǝ ͎͍ Ɣ ͈ Intensive thermodynamic potentials (single component) name independent variables differential form integrated relations entropy per -particle ͧʚ͙, ͪʛ 1 ͊ ͙ ͊ͪ ͧ͘ Ɣ ͙͘ ƍ ͪ͘ Ɣ Ǝͧ ƍ ƍ ͎ ͎ ͎ ͎ ͎ energy per -particle ͙ʚͧ, ͪʛ ͙͘ Ɣ ͎ͧ͘ Ǝ ͊ͪ͘ Ɣ ͙ Ǝ ͎ͧ ƍ ͊ͪ enthalpy per -particle ͜ʚͧ, ͊ʛ ͘͜ Ɣ ͎ͧ͘ ƍ ͪ͊͘ ͜ Ɣ ͙ ƍ ͊ͪ Ɣ ͜ Ǝ ͎ͧ Helmholtz free energy per -particle ͕ʚ͎, ͪʛ ͕͘ Ɣ Ǝ͎ͧ͘ Ǝ ͊ͪ͘ ͕ Ɣ ͙ Ǝ ͎ͧ Ɣ ͕ ƍ ͊ͪ Gibbs free energy per -particle ͛ʚ͎, ͊ʛ ͛͘ Ɣ Ǝ͎ͧ͘ ƍ ͪ͊͘ ͛ Ɣ ͙ ƍ ͊ͪ Ǝ ͎ͧ Ɣ ͕ ƍ ͊ͪ Ɣ ͜ Ǝ ͎ͧ Ɣ ͛ ChE 210A Thermodynamics and statistical mechanics Measurable quantities name definition name definition pressure temperature ͊ ͎ volume heat of phase change constant -volume heat capacity ͐ isothermal compressibility Δ͂ ̿͘ 1 ͐͘ ͘ ln ͐ ̽ Ɣ ƴ Ƹ Ɣ Ǝ ƴ Ƹ Ɣ Ǝ ƴ Ƹ constant -pressure heat capacity ͎͘ thermal expansivity ͐ ͊͘ ͊͘ ͂͘ 1 ͐͘ ͘ ln ͐ ̽ Ɣ ƴ Ƹ Ɣ ƴ Ƹ Ɣ ƴ Ƹ ͎͘ ͐ ͎͘ ͎͘ Thermodynamic calculus manipulations name applies to .. -

Chapter 16 Spontaneity, Entropy, and Free Energy
SpontaneousSpontaneous ProcessesProcesses andand Chapter 16 EntropyEntropy •Thermodynamics lets us predict whether a Spontaneity, process will occur but gives no information Entropy, about the amount of time required for the process. and Free Energy •A spontaneous process is one that occurs without outside intervention. Figure 16.2: The rate of a reaction depends on the pathway First Law of Thermodynamics from reactants to products; • The First Law of Thermodynamics states this is the domain of that the energy of the universe is constant. kinetics. Thermodynamics Ea While energy may change in form (heat, tells us whether a reaction is spontaneous based only work, etc.) and be exchanged between the on the properties of the system & surroundings - the total energy reactants and products. remains constant. To describe the system: The predictions of thermodynamics do not Work done by the system is negative. require knowledge of the Work done on the system is positive. pathway between reactants and products. Heat evolved by the system is negative. Heat absorbed by the system is positive. Enthalpy and Enthalpy Change Enthalpy and Enthalpy Change • In Chapter 6, we tentatively defined • In Chapter 6, we tentatively defined enthalpy in terms of the relationship of ∆H enthalpy in terms of the relationship of ∆H to the heat at constant pressure. to the heat at constant pressure. • This means that at a given temperature and • In practice, we measure certain heats of pressure, a given amount of a substance has a reactions and use them to tabulate enthalpies of o definite enthalpy. formation, ∆H f. • Therefore, if you know the enthalpies of • Standard enthalpies of formation for selected substances, you can calculate the change in compounds are listed in Appendix 4. -

Increasing Temperature Increases Disorder, Because the Entropy Dominates the Free Energy at High Temperatures, Whereas Enthalpy Dominates at Low Temperatures
Increasing temperature increases disorder, because the entropy dominates the free energy at high temperatures, whereas enthalpy dominates at low temperatures. That is, in the expression ΔG = ΔH TΔS, − the entropy term (TΔS) becomes more important (either for ΔS positive or ΔS negative) as T increases; hence at high temperatures the system will become more disordered to increase its entropy, even at the cost of increasing enthalpy. 3. The difference between the energy and enthalpy changes in expanding an ideal gas. How much heat is required to cause the quasi-static isothermal expansion of one mole of an ideal gas at T = 500 K from PA =0.42 atm, VA = 100 liters to PB =0.15 atm? (a) What is VB? (b) What is ΔU for this process? (c) What is ΔH for this process? (a) For an ideal gas, in any isothermal process ΔU = 0. Therefore ΔU = q + w =0 = q = w. ⇒ − For such a process, when it is also quasi-static, V2 nRT V w = pdV = dV = nRT ln 2 , − − V1 V − V1 so V q = nRT ln 2 V1 1 1 V2 = (1 mol)(2 cal K− mol− )(500 K) ln 100 L Using the ideal gas law, V2 p1 p1V1 = nRT = p2V2 = = . ⇒ V1 p2 79 Therefore we can write p q = nRT ln 1 p2 0.42 atm = (1000 cal) ln 0.15 atm = 1030 cal 1000 cal. ≈ (b) From the ideal gas law, p1 0.42 atm V2 = V1 = (100 L) p2 0.15 atm = 280 L. (c) For any isothermal process of an ideal gas, ΔU =0 [ΔU = C (T T )]. -

Maxwell Relations
Maxwell relations Maxwell's relations are a set of equations in thermodynamics which are derivable from the symmetry of second derivatives and from the definitions of the thermodynamic potentials. ese relations are named for the nineteenth- century physicist James Clerk Maxwell. Contents Equations The four most common Maxwell relations Derivation Derivation based on Jacobians General Maxwell relationships See also Equations Flow chart showing the paths between the Maxwell relations. P: e structure of Maxwell relations is a pressure, T: temperature, V: volume, S: entropy, α: coefficient of statement of equality among the second thermal expansion, κ: compressibility, CV: heat capacity at derivatives for continuous functions. It constant volume, CP: heat capacity at constant pressure. follows directly from the fact that the order of differentiation of an analytic function of two variables is irrelevant (Schwarz theorem). In the case of Maxwell relations the function considered is a thermodynamic potential and xi and xj are two different natural variables for that potential: Schwarz' theorem (general) where the partial derivatives are taken with all other natural variables held constant. It is seen that for every thermodynamic potential there are n(n − 1)/2 possible Maxwell relations where n is the number of natural variables for that potential. e substantial increase in the entropy will be verified according to the relations satisfied by the laws of thermodynamics e four most common Maxwell relations e four most common Maxwell relations are the equalities of the second derivatives of each of the four thermodynamic potentials, with respect to their thermal natural variable (temperature T; or entropy S) and their mechanical natural variable (pressure P; or volume V): Maxwell's relations (common) where the potentials as functions of their natural thermal and mechanical variables are the internal energy U(S, V), enthalpy H(S, P), Helmholtz free energy F(T, V) and Gibbs free energy G(T, P).