Enthalpy and Free Energy of Reaction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 12 Lecture Notes
Chemical Kinetics “The study of speeds of reactions and the nanoscale pathways or rearrangements by which atoms and molecules are transformed to products” Chapter 13: Chemical Kinetics: Rates of Reactions © 2008 Brooks/Cole 1 © 2008 Brooks/Cole 2 Reaction Rate Reaction Rate Combustion of Fe(s) powder: © 2008 Brooks/Cole 3 © 2008 Brooks/Cole 4 Reaction Rate Reaction Rate + Change in [reactant] or [product] per unit time. Average rate of the Cv reaction can be calculated: Time, t [Cv+] Average rate + Cresol violet (Cv ; a dye) decomposes in NaOH(aq): (s) (mol / L) (mol L-1 s-1) 0.0 5.000 x 10-5 13.2 x 10-7 10.0 3.680 x 10-5 9.70 x 10-7 20.0 2.710 x 10-5 7.20 x 10-7 30.0 1.990 x 10-5 5.30 x 10-7 40.0 1.460 x 10-5 3.82 x 10-7 + - 50.0 1.078 x 10-5 Cv (aq) + OH (aq) → CvOH(aq) 2.85 x 10-7 60.0 0.793 x 10-5 -7 + + -5 1.82 x 10 change in concentration of Cv Δ [Cv ] 80.0 0.429 x 10 rate = = 0.99 x 10-7 elapsed time Δt 100.0 0.232 x 10-5 © 2008 Brooks/Cole 5 © 2008 Brooks/Cole 6 1 Reaction Rates and Stoichiometry Reaction Rates and Stoichiometry Cv+(aq) + OH-(aq) → CvOH(aq) For any general reaction: a A + b B c C + d D Stoichiometry: The overall rate of reaction is: Loss of 1 Cv+ → Gain of 1 CvOH Rate of Cv+ loss = Rate of CvOH gain 1 Δ[A] 1 Δ[B] 1 Δ[C] 1 Δ[D] Rate = − = − = + = + Another example: a Δt b Δt c Δt d Δt 2 N2O5(g) 4 NO2(g) + O2(g) Reactants decrease with time. -

Organic Chemistry
Wisebridge Learning Systems Organic Chemistry Reaction Mechanisms Pocket-Book WLS www.wisebridgelearning.com © 2006 J S Wetzel LEARNING STRATEGIES CONTENTS ● The key to building intuition is to develop the habit ALKANES of asking how each particular mechanism reflects Thermal Cracking - Pyrolysis . 1 general principles. Look for the concepts behind Combustion . 1 the chemistry to make organic chemistry more co- Free Radical Halogenation. 2 herent and rewarding. ALKENES Electrophilic Addition of HX to Alkenes . 3 ● Acid Catalyzed Hydration of Alkenes . 4 Exothermic reactions tend to follow pathways Electrophilic Addition of Halogens to Alkenes . 5 where like charges can separate or where un- Halohydrin Formation . 6 like charges can come together. When reading Free Radical Addition of HX to Alkenes . 7 organic chemistry mechanisms, keep the elec- Catalytic Hydrogenation of Alkenes. 8 tronegativities of the elements and their valence Oxidation of Alkenes to Vicinal Diols. 9 electron configurations always in your mind. Try Oxidative Cleavage of Alkenes . 10 to nterpret electron movement in terms of energy Ozonolysis of Alkenes . 10 Allylic Halogenation . 11 to make the reactions easier to understand and Oxymercuration-Demercuration . 13 remember. Hydroboration of Alkenes . 14 ALKYNES ● For MCAT preparation, pay special attention to Electrophilic Addition of HX to Alkynes . 15 Hydration of Alkynes. 15 reactions where the product hinges on regio- Free Radical Addition of HX to Alkynes . 16 and stereo-selectivity and reactions involving Electrophilic Halogenation of Alkynes. 16 resonant intermediates, which are special favor- Hydroboration of Alkynes . 17 ites of the test-writers. Catalytic Hydrogenation of Alkynes. 17 Reduction of Alkynes with Alkali Metal/Ammonia . 18 Formation and Use of Acetylide Anion Nucleophiles . -
![Solvent Effects on Structure and Reaction Mechanism: a Theoretical Study of [2 + 2] Polar Cycloaddition Between Ketene and Imine](https://docslib.b-cdn.net/cover/9440/solvent-effects-on-structure-and-reaction-mechanism-a-theoretical-study-of-2-2-polar-cycloaddition-between-ketene-and-imine-159440.webp)
Solvent Effects on Structure and Reaction Mechanism: a Theoretical Study of [2 + 2] Polar Cycloaddition Between Ketene and Imine
J. Phys. Chem. B 1998, 102, 7877-7881 7877 Solvent Effects on Structure and Reaction Mechanism: A Theoretical Study of [2 + 2] Polar Cycloaddition between Ketene and Imine Thanh N. Truong Henry Eyring Center for Theoretical Chemistry, Department of Chemistry, UniVersity of Utah, Salt Lake City, Utah 84112 ReceiVed: March 25, 1998; In Final Form: July 17, 1998 The effects of aqueous solvent on structures and mechanism of the [2 + 2] cycloaddition between ketene and imine were investigated by using correlated MP2 and MP4 levels of ab initio molecular orbital theory in conjunction with the dielectric continuum Generalized Conductor-like Screening Model (GCOSMO) for solvation. We found that reactions in the gas phase and in aqueous solution have very different topology on the free energy surfaces but have similar characteristic motion along the reaction coordinate. First, it involves formation of a planar trans-conformation zwitterionic complex, then a rotation of the two moieties to form the cycloaddition product. Aqueous solvent significantly stabilizes the zwitterionic complex, consequently changing the topology of the free energy surface from a gas-phase single barrier (one-step) process to a double barrier (two-step) one with a stable intermediate. Electrostatic solvent-solute interaction was found to be the dominant factor in lowering the activation energy by 4.5 kcal/mol. The present calculated results are consistent with previous experimental data. Introduction Lim and Jorgensen have also carried out free energy perturbation (FEP) theory simulations with accurate force field that includes + [2 2] cycloaddition reactions are useful synthetic routes to solute polarization effects and found even much larger solvent - formation of four-membered rings. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Eyring Equation
Search Buy/Sell Used Reactors Glass microreactors Hydrogenation Reactor Buy Or Sell Used Reactors Here. Save Time Microreactors made of glass and lab High performance reactor technology Safe And Money Through IPPE! systems for chemical synthesis scale-up. Worldwide supply www.IPPE.com www.mikroglas.com www.biazzi.com Reactors & Calorimeters Induction Heating Reacting Flow Simulation Steam Calculator For Process R&D Laboratories Check Out Induction Heating Software & Mechanisms for Excel steam table add-in for water Automated & Manual Solutions From A Trusted Source. Chemical, Combustion & Materials and steam properties www.helgroup.com myewoss.biz Processes www.chemgoodies.com www.reactiondesign.com Eyring Equation Peter Keusch, University of Regensburg German version "If the Lord Almighty had consulted me before embarking upon the Creation, I should have recommended something simpler." Alphonso X, the Wise of Spain (1223-1284) "Everything should be made as simple as possible, but not simpler." Albert Einstein Both the Arrhenius and the Eyring equation describe the temperature dependence of reaction rate. Strictly speaking, the Arrhenius equation can be applied only to the kinetics of gas reactions. The Eyring equation is also used in the study of solution reactions and mixed phase reactions - all places where the simple collision model is not very helpful. The Arrhenius equation is founded on the empirical observation that rates of reactions increase with temperature. The Eyring equation is a theoretical construct, based on transition state model. The bimolecular reaction is considered by 'transition state theory'. According to the transition state model, the reactants are getting over into an unsteady intermediate state on the reaction pathway. -

Ochem ACS Review 23 Aryl Halides
ACS Review Aryl Halides 1. Which one of the following has the weakest carbon-chlorine bond? A. I B. II C. III D. IV 2. Which compound in each of the following pairs is the most reactive to the conditions indicated? A. I and III B. I and IV C. II and III D. II and IV 3. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. 4-nitrofluorobenzene C. 2,4-dinitrofluorobenzene D. 2,4,6-trinitrofluorobenzene 4. Which of the following reacts at the fastest rate with potassium methoxide (KOCH 3) in methanol? A. fluorobenzene B. p-nitrofluorobenzene C. p-fluorotoluene D. p-bromofluorobenzene 5. Which of the following is the kinetic rate equation for the addition-elimination mechanism of nucleophilic aromatic substitution? A. rate = k[aryl halide] B. rate = k[nucleophile] C. rate = k[aryl halide][nucleophile] D. rate = k[aryl halide][nucleophile] 2 6. Which of the following is not a resonance form of the intermediate in the nucleophilic addition of hydroxide ion to para -fluoronitrobenzene? A. I B. II C. III D. IV 7. Which chlorine is most susceptible to nucleophilic substitution with NaOCH 3 in methanol? A. #1 B. #2 C. #1 and #2 are equally susceptible D. no substitution is possible 8. What is the product of the following reaction? A. N,N-dimethylaniline B. para -chloro-N,N-dimethylaniline C. phenyllithium (C 6H5Li) D. meta -chloro-N,N-dimethylaniline 9. Which one of the reagents readily reacts with bromobenzene without heating? A. -

Estimation of Viscosity Arrhenius Pre-Exponential Factor and Activation Energy of Some Organic Liquids E
ISSN 2350-1030 International Journal of Recent Research in Physics and Chemical Sciences (IJRRPCS) Vol. 5, Issue 1, pp: (18-26), Month: April 2018 – September 2018, Available at: www.paperpublications.org ESTIMATION OF VISCOSITY ARRHENIUS PRE-EXPONENTIAL FACTOR AND ACTIVATION ENERGY OF SOME ORGANIC LIQUIDS E. IKE1 and S. C. EZIKE2 1,2Department of Physics, Modibbo Adama University of Technology, P. M. B. 2076, Yola, Adamawa State, Nigeria. Corresponding Author‟s E-mail: [email protected] Abstract: Information concerning fluid’s physico-chemical behaviors is of utmost importance in the design, running and optimization of industrial processes, in this regard, the idea of fluid viscosity quickly comes to mind. Models have been proposed in literatures to describe the viscosity of liquids and fluids in general. The Arrhenius type equations have been proposed for pure solvents; correlating the pre-exponential factor A and activation energy Ea. In this paper, we aim at extending these Arrhenius parameters to simple organic liquids such as water, ethanol and Diethyl ether. Hence, statistical method and analysis are applied using viscosity data set from the literature of some organic liquids. The Arrhenius type equation simplifies the estimation of viscous properties and the calculations thereafter. Keywords: Viscosity, organic liquids, Arrhenius parameters, correlation, statistics. 1. INTRODUCTION All fluids are compressible and when flowing are capable of sustaining shearing stress on account of friction between the adjacent layers. Viscosity (η) is the inherent property of all fluids and may be referred to as the internal friction offered by a fluid to the flow. For water in a beaker, when stirred and left to itself, the motion subsides after sometime, which can happen only in the presence of resisting force acting on the fluid. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling
Article pubs.acs.org/JCTC Enzymatic Kinetic Isotope Effects from First-Principles Path Sampling Calculations Matthew J. Varga and Steven D. Schwartz* Department of Chemistry and Biochemistry, University of Arizona, Tucson, Arizona 85721, United States *S Supporting Information ABSTRACT: In this study, we develop and test a method to determine the rate of particle transfer and kinetic isotope effects in enzymatic reactions, specifically yeast alcohol dehydrogenase (YADH), from first-principles. Transition path sampling (TPS) and normal mode centroid dynamics (CMD) are used to simulate these enzymatic reactions without knowledge of their reaction coordinates and with the inclusion of quantum effects, such as zero-point energy and tunneling, on the transferring particle. Though previous studies have used TPS to calculate reaction rate constants in various model and real systems, it has not been applied to a system as large as YADH. The calculated primary H/D kinetic isotope effect agrees with previously reported experimental results, within experimental error. The kinetic isotope effects calculated with this method correspond to the kinetic isotope effect of the transfer event itself. The results reported here show that the kinetic isotope effects calculated from first-principles, purely for barrier passage, can be used to predict experimental kinetic isotope effects in enzymatic systems. ■ INTRODUCTION staging method from the Chandler group.10 Another method that uses an energy gap formalism like QCP is a hybrid Particle transfer plays a crucial role in a breadth of enzymatic ff reactions, including hydride, hydrogen atom, electron, and quantum-classical approach of Hammes-Schi er and co-work- 1 ers, which models the transferring particle nucleus as a proton-coupled electron transfer reactions. -

The Arene–Alkene Photocycloaddition
The arene–alkene photocycloaddition Ursula Streit and Christian G. Bochet* Review Open Access Address: Beilstein J. Org. Chem. 2011, 7, 525–542. Department of Chemistry, University of Fribourg, Chemin du Musée 9, doi:10.3762/bjoc.7.61 CH-1700 Fribourg, Switzerland Received: 07 January 2011 Email: Accepted: 23 March 2011 Ursula Streit - [email protected]; Christian G. Bochet* - Published: 28 April 2011 [email protected] This article is part of the Thematic Series "Photocycloadditions and * Corresponding author photorearrangements". Keywords: Guest Editor: A. G. Griesbeck benzene derivatives; cycloadditions; Diels–Alder; photochemistry © 2011 Streit and Bochet; licensee Beilstein-Institut. License and terms: see end of document. Abstract In the presence of an alkene, three different modes of photocycloaddition with benzene derivatives can occur; the [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para photocycloaddition. This short review aims to demonstrate the synthetic power of these photocycloadditions. Introduction Photocycloadditions occur in a variety of modes [1]. The best In the presence of an alkene, three different modes of photo- known representatives are undoubtedly the [2 + 2] photocyclo- cycloaddition with benzene derivatives can occur, viz. the addition, forming either cyclobutanes or four-membered hetero- [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para cycles (as in the Paternò–Büchi reaction), whilst excited-state photocycloaddition (Scheme 2). The descriptors ortho, meta [4 + 4] cycloadditions can also occur to afford cyclooctadiene and para only indicate the connectivity to the aromatic ring, and compounds. On the other hand, the well-known thermal [4 + 2] do not have any implication with regard to the reaction mecha- cycloaddition (Diels–Alder reaction) is only very rarely nism. -
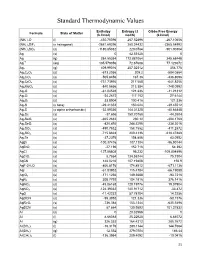
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems Nathan Mitchell East Tennessee State University
East Tennessee State University Digital Commons @ East Tennessee State University Electronic Theses and Dissertations Student Works 5-2018 Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems Nathan Mitchell East Tennessee State University Follow this and additional works at: https://dc.etsu.edu/etd Part of the Analytical Chemistry Commons Recommended Citation Mitchell, Nathan, "Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems" (2018). Electronic Theses and Dissertations. Paper 3430. https://dc.etsu.edu/etd/3430 This Thesis - Open Access is brought to you for free and open access by the Student Works at Digital Commons @ East Tennessee State University. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ East Tennessee State University. For more information, please contact [email protected]. Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems ________________________ A thesis presented to the faculty of the Department of Chemistry East Tennessee State University In partial fulfillment of the requirements for the degree Master of Science in Chemistry ______________________ by Nathan Mitchell May 2018 _____________________ Dr. Dane Scott, Chair Dr. Greg Bishop Dr. Marina Roginskaya Keywords: Thermodynamic Analysis, Hydrolysis, Linear Solvent Energy Relationships, Cosolvent Systems, Acetonitrile ABSTRACT Eyring Activation Energy Analysis of Acetic Anhydride Hydrolysis in Acetonitrile Cosolvent Systems by Nathan Mitchell Acetic anhydride hydrolysis in water is considered a standard reaction for investigating activation energy parameters using cosolvents. Hydrolysis in water/acetonitrile cosolvent is monitored by measuring pH vs. time at temperatures from 15.0 to 40.0 °C and mole fraction of water from 1 to 0.750.