Thermodynamic Laws & Gibbs Free Energy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ENERGY, ENTROPY, and INFORMATION Jean Thoma June
ENERGY, ENTROPY, AND INFORMATION Jean Thoma June 1977 Research Memoranda are interim reports on research being conducted by the International Institute for Applied Systems Analysis, and as such receive only limited scientific review. Views or opinions contained herein do not necessarily represent those of the Institute or of the National Member Organizations supporting the Institute. PREFACE This Research Memorandum contains the work done during the stay of Professor Dr.Sc. Jean Thoma, Zug, Switzerland, at IIASA in November 1976. It is based on extensive discussions with Professor HAfele and other members of the Energy Program. Al- though the content of this report is not yet very uniform because of the different starting points on the subject under consideration, its publication is considered a necessary step in fostering the related discussion at IIASA evolving around th.e problem of energy demand. ABSTRACT Thermodynamical considerations of energy and entropy are being pursued in order to arrive at a general starting point for relating entropy, negentropy, and information. Thus one hopes to ultimately arrive at a common denominator for quanti- ties of a more general nature, including economic parameters. The report closes with the description of various heating appli- cation.~and related efficiencies. Such considerations are important in order to understand in greater depth the nature and composition of energy demand. This may be highlighted by the observation that it is, of course, not the energy that is consumed or demanded for but the informa- tion that goes along with it. TABLE 'OF 'CONTENTS Introduction ..................................... 1 2 . Various Aspects of Entropy ........................2 2.1 i he no me no logical Entropy ........................ -

31 Jan 2021 Laws of Thermodynamics . L01–1 Review of Thermodynamics. 1
31 jan 2021 laws of thermodynamics . L01{1 Review of Thermodynamics. 1: The Basic Laws What is Thermodynamics? • Idea: The study of states of physical systems that can be characterized by macroscopic variables, usu- ally equilibrium states (mechanical, thermal or chemical), and possible transformations between them. It started as a phenomenological subject motivated by practical applications, but it gradually developed into a coherent framework that we will view here as the macroscopic counterpart to the statistical mechanics of the microscopic constituents, and provides the observational context in which to verify many of its predictions. • Plan: We will mostly be interested in internal states, so the allowed processes will include heat exchanges and the main variables will always include the internal energy and temperature. We will recall the main facts (definitions, laws and relationships) of thermodynamics and discuss physical properties that characterize different substances, rather than practical applications such as properties of specific engines. The connection with statistical mechanics, based on a microscopic model of each system, will be established later. States and State Variables for a Thermodynamical System • Energy: The internal energy E is the central quantity in the theory, and is seen as a function of some complete set of variables characterizing each state. Notice that often energy is the only relevant macroscopic conservation law, while momentum, angular momentum or other quantities may not need to be considered. • Extensive variables: For each system one can choose a set of extensive variables (S; X~ ) whose values specify ~ the equilibrium states of the system; S is the entropy and the X are quantities that may include V , fNig, ~ ~ q, M, ~p, L, .. -

Lecture 4: 09.16.05 Temperature, Heat, and Entropy
3.012 Fundamentals of Materials Science Fall 2005 Lecture 4: 09.16.05 Temperature, heat, and entropy Today: LAST TIME .........................................................................................................................................................................................2� State functions ..............................................................................................................................................................................2� Path dependent variables: heat and work..................................................................................................................................2� DEFINING TEMPERATURE ...................................................................................................................................................................4� The zeroth law of thermodynamics .............................................................................................................................................4� The absolute temperature scale ..................................................................................................................................................5� CONSEQUENCES OF THE RELATION BETWEEN TEMPERATURE, HEAT, AND ENTROPY: HEAT CAPACITY .......................................6� The difference between heat and temperature ...........................................................................................................................6� Defining heat capacity.................................................................................................................................................................6� -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Thermodynamics, Flame Temperature and Equilibrium
Thermodynamics, Flame Temperature and Equilibrium Combustion Summer School 2018 Prof. Dr.-Ing. Heinz Pitsch Course Overview Part I: Fundamentals and Laminar Flames • Introduction • Fundamentals and mass balances of combustion systems • Thermodynamic quantities • Thermodynamics, flame • Flame temperature at complete temperature, and equilibrium conversion • Governing equations • Chemical equilibrium • Laminar premixed flames: Kinematics and burning velocity • Laminar premixed flames: Flame structure • Laminar diffusion flames • FlameMaster flame calculator 2 Thermodynamic Quantities First law of thermodynamics - balance between different forms of energy • Change of specific internal energy: du specific work due to volumetric changes: δw = -pdv , v=1/ρ specific heat transfer from the surroundings: δq • Related quantities specific enthalpy (general definition): specific enthalpy for an ideal gas: • Energy balance for a closed system: 3 Multicomponent system • Specific internal energy and specific enthalpy of mixtures • Relation between internal energy and enthalpy of single species 4 Multicomponent system • Ideal gas u and h only function of temperature • If cpi is specific heat at constant pressure and hi,ref is reference enthalpy at reference temperature Tref , temperature dependence of partial specific enthalpy is given by • Reference temperature may be arbitrarily chosen, most frequently used: Tref = 0 K or Tref = 298.15 K 5 Multicomponent system • Partial molar enthalpy hi,m is and its temperature dependence is where the molar specific -

A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Article A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action Ivan Kennedy 1,2,*, Harold Geering 2, Michael Rose 3 and Angus Crossan 2 1 Sydney Institute of Agriculture, University of Sydney, NSW 2006, Australia 2 QuickTest Technologies, PO Box 6285 North Ryde, NSW 2113, Australia; [email protected] (H.G.); [email protected] (A.C.) 3 NSW Department of Primary Industries, Wollongbar NSW 2447, Australia; [email protected] * Correspondence: [email protected]; Tel.: + 61-4-0794-9622 Received: 23 March 2019; Accepted: 26 April 2019; Published: 1 May 2019 Abstract: A convenient practical model for accurately estimating the total entropy (ΣSi) of atmospheric gases based on physical action is proposed. This realistic approach is fully consistent with statistical mechanics, but reinterprets its partition functions as measures of translational, rotational, and vibrational action or quantum states, to estimate the entropy. With all kinds of molecular action expressed as logarithmic functions, the total heat required for warming a chemical system from 0 K (ΣSiT) to a given temperature and pressure can be computed, yielding results identical with published experimental third law values of entropy. All thermodynamic properties of gases including entropy, enthalpy, Gibbs energy, and Helmholtz energy are directly estimated using simple algorithms based on simple molecular and physical properties, without resource to tables of standard values; both free energies are measures of quantum field states and of minimal statistical degeneracy, decreasing with temperature and declining density. We propose that this more realistic approach has heuristic value for thermodynamic computation of atmospheric profiles, based on steady state heat flows equilibrating with gravity. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

Nonequilibrium Thermodynamics and Scale Invariance
Article Nonequilibrium Thermodynamics and Scale Invariance Leonid M. Martyushev 1,2,* and Vladimir Celezneff 3 1 Technical Physics Department, Ural Federal University, 19 Mira St., Ekaterinburg 620002, Russia 2 Institute of Industrial Ecology, Russian Academy of Sciences, 20 S. Kovalevskaya St., Ekaterinburg 620219, Russia 3 The Racah Institute of Physics, Hebrew University of Jerusalem, Jerusalem 91904, Israel; [email protected] * Correspondence: [email protected]; Tel.: +7-92-222-77-425 Academic Editors: Milivoje M. Kostic, Miguel Rubi and Kevin H. Knuth Received: 30 January 2017; Accepted: 14 March 2017; Published: 16 March 2017 Abstract: A variant of continuous nonequilibrium thermodynamic theory based on the postulate of the scale invariance of the local relation between generalized fluxes and forces is proposed here. This single postulate replaces the assumptions on local equilibrium and on the known relation between thermodynamic fluxes and forces, which are widely used in classical nonequilibrium thermodynamics. It is shown here that such a modification not only makes it possible to deductively obtain the main results of classical linear nonequilibrium thermodynamics, but also provides evidence for a number of statements for a nonlinear case (the maximum entropy production principle, the macroscopic reversibility principle, and generalized reciprocity relations) that are under discussion in the literature. Keywords: dissipation function; nonequilibrium thermodynamics; generalized fluxes and forces 1. Introduction Classical nonequilibrium thermodynamics is an important field of modern physics that was developed for more than a century [1–5]. It is fundamentally based on thermodynamics and statistical physics (including kinetic theory and theory of random processes) and is widely applied in biophysics, geophysics, chemistry, economics, etc. -

Ch. 3. the Third Law of Thermodynamics (Pdf)
3-1 CHAPTER 3 THE THIRD LAW OF THERMODYNAMICS1 In sharp contrast to the first two laws, the third law of thermodynamics can be characterized by diverse expression2, disputed descent, and questioned authority.3 Since first advanced by Nernst4 in 1906 as the Heat Theorem, its thermodynamic status has been controversial; its usefulness, however, is unquestioned. 3.1 THE HEAT THEOREM The Heat Theorem was first proposed as an empirical generalization based on the temperature dependence of the internal energy change, ∆U, and the Helmholtz free energy change, ∆A, for chemical reactions involving condensed phases. As the absolute temperature, T, approaches zero, ∆U and ∆A by definition become equal, but The Heat Theorem stated that d∆U/dT and d∆A/dT also approach zero. These derivatives are ∆Cv and -∆S respectively. The statement that ∆Cv equals zero would attract little attention today in view of the abundance of experimental and theoretical evidence showing that the heat capacities of condensed phases approach zero as zero absolute temperature is approached. However, even today the controversial and enigmatic aspect of The Heat Theorem is the equivalent statement 1 Most of this chapter is taken from B.G. Kyle, Chem. Eng. Ed., 28(3), 176 (1994). 2 For a sampling of expressions see E. M. Loebl, J. Chem. Educ., 37, 361 (1960). 3 For extreme positions see E. D. Eastman, Chem. Rev., 18, 257 (1936). 4 All of Nernst's work in this area is covered in W. Nernst, The New Heat Theorem; Dutton: New York, 1926. 3-2 lim ∆S = 0 (3-1) T → 0 In 1912 Nernst offered a proof that the unattainability of zero absolute temperature was dictated by the second law of thermodynamics and was able to show that Eq. -
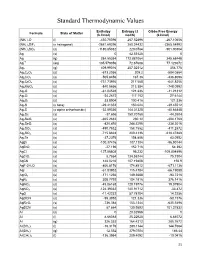
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

Bsc Chemistry
Subject Chemistry Paper No and Title 10: Physical Chemistry –III (Classical Thermodynamics, Non-Equilibrium Thermodynamics, Surface Chemistry, Fast Kinetics) Module No and Title 12: Thermodynamic criteria for non-equilibrium states Module Tag CHE_P10_M12 CHEMISTRY Paper No. 10: Physical Chemistry –III (Classical Thermodynamics, Non-Equilibrium Thermodynamics, Surface Chemistry, Fast Kinetics) Module No. 12: Thermodynamic criteria for nonequilibrium states TABLE OF CONTENTS 1. Learning Outcomes 2. Introduction 3. Difference between equilibrium and non-equilibrium thermodynamics 4. Postulates of irreversible thermodynamics 5. Non equilibrium state variables 6. Basic concepts 6.1 Zeroth law of thermodynamics 6.2 First law of thermodynamics 6.3 Second law of thermodynamics 6.4 Third law of thermodynamics 7. Summary CHEMISTRY Paper No. 10: Physical Chemistry –III (Classical Thermodynamics, Non-Equilibrium Thermodynamics, Surface Chemistry, Fast Kinetics) Module No. 12: Thermodynamic criteria for nonequilibrium states 1. Learning Outcomes After studying this module you shall be able to: Know the significance of irreversible thermodynamics Differentiate between equilibrium and non-equilibrium thermodynamics. Learn postulates of non-equilibrium thermodynamics. Know about different laws of thermodynamics (first, second and third) laws. 2. Introduction In classical thermodynamics, we have seen time dependent variation of thermodynamic quantities such as is internal energy (U), enthalpy (H), entropy (S),Gibb’s free energy (G) etc. are not considered. Classical thermodynamics deals with transitions from one equilibrium state to another brought about by different mechanical or chemical methods. Non equilibrium thermodynamics is that branch of thermodynamics that deals with the system which are not in thermodynamic equilibrium. But such systems can be described by non-equilibrium state variables which represent an extrapolation of variables used to specify system in thermodynamic equilibrium. -

Lecture 6: Entropy
Matthew Schwartz Statistical Mechanics, Spring 2019 Lecture 6: Entropy 1 Introduction In this lecture, we discuss many ways to think about entropy. The most important and most famous property of entropy is that it never decreases Stot > 0 (1) Here, Stot means the change in entropy of a system plus the change in entropy of the surroundings. This is the second law of thermodynamics that we met in the previous lecture. There's a great quote from Sir Arthur Eddington from 1927 summarizing the importance of the second law: If someone points out to you that your pet theory of the universe is in disagreement with Maxwell's equationsthen so much the worse for Maxwell's equations. If it is found to be contradicted by observationwell these experimentalists do bungle things sometimes. But if your theory is found to be against the second law of ther- modynamics I can give you no hope; there is nothing for it but to collapse in deepest humiliation. Another possibly relevant quote, from the introduction to the statistical mechanics book by David Goodstein: Ludwig Boltzmann who spent much of his life studying statistical mechanics, died in 1906, by his own hand. Paul Ehrenfest, carrying on the work, died similarly in 1933. Now it is our turn to study statistical mechanics. There are many ways to dene entropy. All of them are equivalent, although it can be hard to see. In this lecture we will compare and contrast dierent denitions, building up intuition for how to think about entropy in dierent contexts. The original denition of entropy, due to Clausius, was thermodynamic.