A Simple Method to Estimate Entropy and Free Energy of Atmospheric Gases from Their Action
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DFT Quantization of the Gibbs Free Energy of a Quantum Body
DFT quantization of the Gibbs Free Energy of a quantum body S. Selenu (Dated: December 2, 2020) In this article it is introduced a theoretical model made in order to perform calculations of the quantum heat of a body that could be acquired or delivered during a thermal transformation of its quantum states. Here the model is mainly targeted to the electronic structure of matter[1] at the nano and micro scale where DFT models have been frequently developed of the total energy of quantum systems. De fining an Entropy functional S[ρ] makes us able optimizing a free energy G[ρ] of the quantum system at finite temperatures. Due to the generality of the model, the latter can be also applied to several first principles computational codes where ab initio modelling of quantum matter is asked. INTRODUCTION thermal transformations at a fi nite temperature T . This work could then be consid- This article will be focused on a study based on the ered as the begining part of a more general theory can derivation of the quantum electrical heat absorbed or ei- include an ab initio modelling of the first law and second ther released by a physical system made of atoms and law of thermodynamics[13], where heat and free energy electrons either at the nano or at the microscopic scale, variations of a quantum body are taken into account. In extending to it the concept of thermal heat within a DFT fact it is actually considered a quantum system of elec- model. Also, the latter being fundamental in physics, can trons at a given temperature T in thermal equilibrium be employed for a full understanding of the thermal phase with its surrounding. -

ENERGY, ENTROPY, and INFORMATION Jean Thoma June
ENERGY, ENTROPY, AND INFORMATION Jean Thoma June 1977 Research Memoranda are interim reports on research being conducted by the International Institute for Applied Systems Analysis, and as such receive only limited scientific review. Views or opinions contained herein do not necessarily represent those of the Institute or of the National Member Organizations supporting the Institute. PREFACE This Research Memorandum contains the work done during the stay of Professor Dr.Sc. Jean Thoma, Zug, Switzerland, at IIASA in November 1976. It is based on extensive discussions with Professor HAfele and other members of the Energy Program. Al- though the content of this report is not yet very uniform because of the different starting points on the subject under consideration, its publication is considered a necessary step in fostering the related discussion at IIASA evolving around th.e problem of energy demand. ABSTRACT Thermodynamical considerations of energy and entropy are being pursued in order to arrive at a general starting point for relating entropy, negentropy, and information. Thus one hopes to ultimately arrive at a common denominator for quanti- ties of a more general nature, including economic parameters. The report closes with the description of various heating appli- cation.~and related efficiencies. Such considerations are important in order to understand in greater depth the nature and composition of energy demand. This may be highlighted by the observation that it is, of course, not the energy that is consumed or demanded for but the informa- tion that goes along with it. TABLE 'OF 'CONTENTS Introduction ..................................... 1 2 . Various Aspects of Entropy ........................2 2.1 i he no me no logical Entropy ........................ -
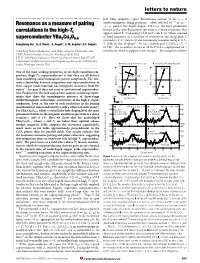
Resonance As a Measure of Pairing Correlations in the High-Tc
letters to nature ................................................................. lost while magnetic (spin) ¯uctuations centred at the x =0 6±11 Resonance as a measure of pairing antiferromagnetic Bragg positionsÐoften referred to as Q0 = (p, p)±persist. For highly doped (123)O6+x, the most prominent feature in the spin ¯uctuation spectrum is a sharp resonance that correlations in the high-Tc appears below Tc at an energy of 41 meV (refs 6±8). When scanned superconductor YBa Cu O at ®xed frequency as a function of wavevector, the sharp peak is 2 3 6.6 centred at (p, p) (refs 6±8) and its intensity is unaffected by a 11.5- 19 Pengcheng Dai*, H. A. Mook*, G. Aeppli², S. M. Hayden³ & F. DogÏan§ T ®eld in the ab-plane . In our underdoped (123)O6.6 (Tc = 62.7 K)9, the resonance occurs at 34 meV and is superposed on a * Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831-6393, USA continuum which is gapped at low energies11. For frequencies below ² NEC Research Institute, Princeton, New Jersey 08540, USA ³ H. H. Wills Physics Laboratory, University of Bristol, Bristol BS8 1TL, UK § Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA B rotated 20.6° from the c-axis B along the [3H,-H,0] direction 2.0 2.0 .............................................................................................................................................. a d [H,3H,0] One of the most striking properties of the high-transition-tem- 1.5 1.5 perature (high-Tc) superconductors is that they are all derived [H, (5-H)/3, 0] from insulating antiferromagnetic parent compounds. The inti- 1.0 B ∼ i c-axis 1.0 B i ab-plane mate relationship between magnetism and superconductivity in these copper oxide materials has intrigued researchers from the [0,K,0] (r.l.u.) [0,K,0] (r.l.u.) 0.5 0.5 outset1±4, because it does not exist in conventional superconduc- 20.6° tors. -

Lecture 4: 09.16.05 Temperature, Heat, and Entropy
3.012 Fundamentals of Materials Science Fall 2005 Lecture 4: 09.16.05 Temperature, heat, and entropy Today: LAST TIME .........................................................................................................................................................................................2� State functions ..............................................................................................................................................................................2� Path dependent variables: heat and work..................................................................................................................................2� DEFINING TEMPERATURE ...................................................................................................................................................................4� The zeroth law of thermodynamics .............................................................................................................................................4� The absolute temperature scale ..................................................................................................................................................5� CONSEQUENCES OF THE RELATION BETWEEN TEMPERATURE, HEAT, AND ENTROPY: HEAT CAPACITY .......................................6� The difference between heat and temperature ...........................................................................................................................6� Defining heat capacity.................................................................................................................................................................6� -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

LECTURES on MATHEMATICAL STATISTICAL MECHANICS S. Adams
ISSN 0070-7414 Sgr´ıbhinn´ıInstiti´uid Ard-L´einnBhaile´ Atha´ Cliath Sraith. A. Uimh 30 Communications of the Dublin Institute for Advanced Studies Series A (Theoretical Physics), No. 30 LECTURES ON MATHEMATICAL STATISTICAL MECHANICS By S. Adams DUBLIN Institi´uid Ard-L´einnBhaile´ Atha´ Cliath Dublin Institute for Advanced Studies 2006 Contents 1 Introduction 1 2 Ergodic theory 2 2.1 Microscopic dynamics and time averages . .2 2.2 Boltzmann's heuristics and ergodic hypothesis . .8 2.3 Formal Response: Birkhoff and von Neumann ergodic theories9 2.4 Microcanonical measure . 13 3 Entropy 16 3.1 Probabilistic view on Boltzmann's entropy . 16 3.2 Shannon's entropy . 17 4 The Gibbs ensembles 20 4.1 The canonical Gibbs ensemble . 20 4.2 The Gibbs paradox . 26 4.3 The grandcanonical ensemble . 27 4.4 The "orthodicity problem" . 31 5 The Thermodynamic limit 33 5.1 Definition . 33 5.2 Thermodynamic function: Free energy . 37 5.3 Equivalence of ensembles . 42 6 Gibbs measures 44 6.1 Definition . 44 6.2 The one-dimensional Ising model . 47 6.3 Symmetry and symmetry breaking . 51 6.4 The Ising ferromagnet in two dimensions . 52 6.5 Extreme Gibbs measures . 57 6.6 Uniqueness . 58 6.7 Ergodicity . 60 7 A variational characterisation of Gibbs measures 62 8 Large deviations theory 68 8.1 Motivation . 68 8.2 Definition . 70 8.3 Some results for Gibbs measures . 72 i 9 Models 73 9.1 Lattice Gases . 74 9.2 Magnetic Models . 75 9.3 Curie-Weiss model . 77 9.4 Continuous Ising model . -

Entropy and H Theorem the Mathematical Legacy of Ludwig Boltzmann
ENTROPY AND H THEOREM THE MATHEMATICAL LEGACY OF LUDWIG BOLTZMANN Cambridge/Newton Institute, 15 November 2010 C´edric Villani University of Lyon & Institut Henri Poincar´e(Paris) Cutting-edge physics at the end of nineteenth century Long-time behavior of a (dilute) classical gas Take many (say 1020) small hard balls, bouncing against each other, in a box Let the gas evolve according to Newton’s equations Prediction by Maxwell and Boltzmann The distribution function is asymptotically Gaussian v 2 f(t, x, v) a exp | | as t ≃ − 2T → ∞ Based on four major conceptual advances 1865-1875 Major modelling advance: Boltzmann equation • Major mathematical advance: the statistical entropy • Major physical advance: macroscopic irreversibility • Major PDE advance: qualitative functional study • Let us review these advances = journey around centennial scientific problems ⇒ The Boltzmann equation Models rarefied gases (Maxwell 1865, Boltzmann 1872) f(t, x, v) : density of particles in (x, v) space at time t f(t, x, v) dxdv = fraction of mass in dxdv The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution f(t, x, v): ∂f 3 ∂f + v = Q(f,f) = ∂t i ∂x Xi=1 i ′ ′ B(v v∗,σ) f(t, x, v )f(t, x, v∗) f(t, x, v)f(t, x, v∗) dv∗ dσ R3 2 − − Z v∗ ZS h i The Boltzmann equation (without boundaries) Unknown = time-dependent distribution -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

A Molecular Modeler's Guide to Statistical Mechanics
A Molecular Modeler’s Guide to Statistical Mechanics Course notes for BIOE575 Daniel A. Beard Department of Bioengineering University of Washington Box 3552255 [email protected] (206) 685 9891 April 11, 2001 Contents 1 Basic Principles and the Microcanonical Ensemble 2 1.1 Classical Laws of Motion . 2 1.2 Ensembles and Thermodynamics . 3 1.2.1 An Ensembles of Particles . 3 1.2.2 Microscopic Thermodynamics . 4 1.2.3 Formalism for Classical Systems . 7 1.3 Example Problem: Classical Ideal Gas . 8 1.4 Example Problem: Quantum Ideal Gas . 10 2 Canonical Ensemble and Equipartition 15 2.1 The Canonical Distribution . 15 2.1.1 A Derivation . 15 2.1.2 Another Derivation . 16 2.1.3 One More Derivation . 17 2.2 More Thermodynamics . 19 2.3 Formalism for Classical Systems . 20 2.4 Equipartition . 20 2.5 Example Problem: Harmonic Oscillators and Blackbody Radiation . 21 2.5.1 Classical Oscillator . 22 2.5.2 Quantum Oscillator . 22 2.5.3 Blackbody Radiation . 23 2.6 Example Application: Poisson-Boltzmann Theory . 24 2.7 Brief Introduction to the Grand Canonical Ensemble . 25 3 Brownian Motion, Fokker-Planck Equations, and the Fluctuation-Dissipation Theo- rem 27 3.1 One-Dimensional Langevin Equation and Fluctuation- Dissipation Theorem . 27 3.2 Fokker-Planck Equation . 29 3.3 Brownian Motion of Several Particles . 30 3.4 Fluctuation-Dissipation and Brownian Dynamics . 32 1 Chapter 1 Basic Principles and the Microcanonical Ensemble The first part of this course will consist of an introduction to the basic principles of statistical mechanics (or statistical physics) which is the set of theoretical techniques used to understand microscopic systems and how microscopic behavior is reflected on the macroscopic scale. -

Entropy: from the Boltzmann Equation to the Maxwell Boltzmann Distribution
Entropy: From the Boltzmann equation to the Maxwell Boltzmann distribution A formula to relate entropy to probability Often it is a lot more useful to think about entropy in terms of the probability with which different states are occupied. Lets see if we can describe entropy as a function of the probability distribution between different states. N! WN particles = n1!n2!....nt! stirling (N e)N N N W = = N particles n1 n2 nt n1 n2 nt (n1 e) (n2 e) ....(nt e) n1 n2 ...nt with N pi = ni 1 W = N particles n1 n2 nt p1 p2 ...pt takeln t lnWN particles = "#ni ln pi i=1 divide N particles t lnW1particle = "# pi ln pi i=1 times k t k lnW1particle = "k# pi ln pi = S1particle i=1 and t t S = "N k p ln p = "R p ln p NA A # i # i i i=1 i=1 ! Think about how this equation behaves for a moment. If any one of the states has a probability of occurring equal to 1, then the ln pi of that state is 0 and the probability of all the other states has to be 0 (the sum over all probabilities has to be 1). So the entropy of such a system is 0. This is exactly what we would have expected. In our coin flipping case there was only one way to be all heads and the W of that configuration was 1. Also, and this is not so obvious, the maximal entropy will result if all states are equally populated (if you want to see a mathematical proof of this check out Ken Dill’s book on page 85). -

Proximity to a Critical Point Driven by Electronic Entropy in Uru2si2
www.nature.com/npjquantmats ARTICLE OPEN Proximity to a critical point driven by electronic entropy in URu2Si2 ✉ Neil Harrison 1 , Satya K. Kushwaha1,2, Mun K. Chan 1 and Marcelo Jaime 1 The strongly correlated actinide metal URu2Si2 exhibits a mean field-like second order phase transition at To ≈ 17 K, yet lacks definitive signatures of a broken symmetry. Meanwhile, various experiments have also shown the electronic energy gap to closely resemble that resulting from hybridization between conduction electron and 5f-electron states. We argue here, using thermodynamic measurements, that the above seemingly incompatible observations can be jointly understood by way of proximity to an entropy-driven critical point, in which the latent heat of a valence-type electronic instability is quenched by thermal excitations across a gap, driving the transition second order. Salient features of such a transition include a robust gap spanning highly degenerate features in the electronic density of states, that is weakly (if at all) suppressed by temperature on approaching To, and an elliptical phase boundary in magnetic field and temperature that is Pauli paramagnetically limited at its critical magnetic field. npj Quantum Materials (2021) 6:24 ; https://doi.org/10.1038/s41535-021-00317-6 1234567890():,; INTRODUCTION no absolute requirement for the magnitude of a hybridization gap URu2Si2 remains of immense interest owing to the possibility to vanish at a phase transition, it need not be thermally of it exhibiting a form of order distinct from that observed -

Boltzmann Equation II: Binary Collisions
Physics 127b: Statistical Mechanics Boltzmann Equation II: Binary Collisions Binary collisions in a classical gas Scattering out Scattering in v'1 v'2 v 1 v 2 R v 2 v 1 v'2 v'1 Center of V' Mass Frame V θ θ b sc sc R V V' Figure 1: Binary collisions in a gas: top—lab frame; bottom—centre of mass frame Binary collisions in a gas are very similar, except that the scattering is off another molecule. An individual scattering process is, of course, simplest to describe in the center of mass frame in terms of the relative E velocity V =Ev1 −Ev2. However the center of mass frame is different for different collisions, so we must keep track of the results in the lab frame, and this makes the calculation rather intricate. I will indicate the main ideas here, and refer you to Reif or Landau and Lifshitz for precise discussions. Lets first set things up in the lab frame. Again we consider the pair of scattering in and scattering out processes that are space-time inverses, and so have identical cross sections. We can write abstractly for the scattering out from velocity vE1 due to collisions with molecules with all velocities vE2, which will clearly be proportional to the number f (vE1) of molecules at vE1 (which we write as f1—sorry, not the same notation as in the previous sections where f1 denoted the deviation of f from the equilibrium distribution!) and the 3 3 number f2d v2 ≡ f(vE2)d v2 in each velocity volume element, and then we must integrate over all possible vE0 vE0 outgoing velocities 1 and 2 ZZZ df (vE ) 1 =− w(vE0 , vE0 ;Ev , vE )f f d3v d3v0 d3v0 .