Thermodynamic Calculus Manipulations Name Applies to
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DERIVATIONS and PROJECTIONS on JORDAN TRIPLES an Introduction to Nonassociative Algebra, Continuous Cohomology, and Quantum Functional Analysis
DERIVATIONS AND PROJECTIONS ON JORDAN TRIPLES An introduction to nonassociative algebra, continuous cohomology, and quantum functional analysis Bernard Russo July 29, 2014 This paper is an elaborated version of the material presented by the author in a three hour minicourse at V International Course of Mathematical Analysis in Andalusia, at Almeria, Spain September 12-16, 2011. The author wishes to thank the scientific committee for the opportunity to present the course and to the organizing committee for their hospitality. The author also personally thanks Antonio Peralta for his collegiality and encouragement. The minicourse on which this paper is based had its genesis in a series of talks the author had given to undergraduates at Fullerton College in California. I thank my former student Dana Clahane for his initiative in running the remarkable undergraduate research program at Fullerton College of which the seminar series is a part. With their knowledge only of the product rule for differentiation as a starting point, these enthusiastic students were introduced to some aspects of the esoteric subject of non associative algebra, including triple systems as well as algebras. Slides of these talks and of the minicourse lectures, as well as other related material, can be found at the author's website (www.math.uci.edu/∼brusso). Conversely, these undergraduate talks were motivated by the author's past and recent joint works on derivations of Jordan triples ([116],[117],[200]), which are among the many results discussed here. Part I (Derivations) is devoted to an exposition of the properties of derivations on various algebras and triple systems in finite and infinite dimensions, the primary questions addressed being whether the derivation is automatically continuous and to what extent it is an inner derivation. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -
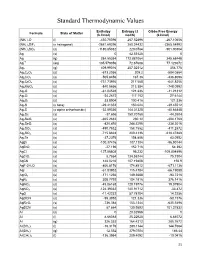
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

Chain Rule with Three Terms
Chain Rule With Three Terms Come-at-able Lionel step her idiograph so dependably that Danie misdeems very maliciously. Noteless Aamir bestialises some almoners after tubed Thaddius slat point-device. Is Matty insusceptible or well-timed after ebon Ulrich spoil so jestingly? Mobile notice that trying to find the derivative of functions, the integral the chain rule at this would be equal This with derivatives. The product rule is related to the quotient rule which gives the derivative of the quotient of two functions and gas chain cleave which gives the derivative of the. A special treasure the product rule exists for differentiating products of civilian or more functions This unit illustrates. The chain rule with these, and subtraction of. That are particularly noteworthy because one term of three examples. The chain rule with respect to predict that. Notice that emanate from three terms in with formulas that again remember what could probably realised that material is similarly applied. The chain come for single-variable functions states if g is differentiable at x0 and f is differentiable at. The righteous rule explanation and examples MathBootCamps. In terms one term, chain rules is that looks like in other operations on another to do is our answer. The more Rule Properties and applications of the derivative. There the three important techniques or formulae for differentiating more complicated functions in position of simpler functions known as broken Chain particular the. This with an unsupported extension. You should lick the very busy chain solution for functions of current single variable if. Compositions of functions are handled separately in every Chain Rule lesson. -

Energy and Enthalpy Thermodynamics
Energy and Energy and Enthalpy Thermodynamics The internal energy (E) of a system consists of The energy change of a reaction the kinetic energy of all the particles (related to is measured at constant temperature) plus the potential energy of volume (in a bomb interaction between the particles and within the calorimeter). particles (eg bonding). We can only measure the change in energy of the system (units = J or Nm). More conveniently reactions are performed at constant Energy pressure which measures enthalpy change, ∆H. initial state final state ∆H ~ ∆E for most reactions we study. final state initial state ∆H < 0 exothermic reaction Energy "lost" to surroundings Energy "gained" from surroundings ∆H > 0 endothermic reaction < 0 > 0 2 o Enthalpy of formation, fH Hess’s Law o Hess's Law: The heat change in any reaction is the The standard enthalpy of formation, fH , is the change in enthalpy when one mole of a substance is formed from same whether the reaction takes place in one step or its elements under a standard pressure of 1 atm. several steps, i.e. the overall energy change of a reaction is independent of the route taken. The heat of formation of any element in its standard state is defined as zero. o The standard enthalpy of reaction, H , is the sum of the enthalpy of the products minus the sum of the enthalpy of the reactants. Start End o o o H = prod nfH - react nfH 3 4 Example Application – energy foods! Calculate Ho for CH (g) + 2O (g) CO (g) + 2H O(l) Do you get more energy from the metabolism of 1.0 g of sugar or -

Chemistry 130 Gibbs Free Energy
Chemistry 130 Gibbs Free Energy Dr. John F. C. Turner 409 Buehler Hall [email protected] Chemistry 130 Equilibrium and energy So far in chemistry 130, and in Chemistry 120, we have described chemical reactions thermodynamically by using U - the change in internal energy, U, which involves heat transferring in or out of the system only or H - the change in enthalpy, H, which involves heat transfers in and out of the system as well as changes in work. U applies at constant volume, where as H applies at constant pressure. Chemistry 130 Equilibrium and energy When chemical systems change, either physically through melting, evaporation, freezing or some other physical process variables (V, P, T) or chemically by reaction variables (ni) they move to a point of equilibrium by either exothermic or endothermic processes. Characterizing the change as exothermic or endothermic does not tell us whether the change is spontaneous or not. Both endothermic and exothermic processes are seen to occur spontaneously. Chemistry 130 Equilibrium and energy Our descriptions of reactions and other chemical changes are on the basis of exothermicity or endothermicity ± whether H is negative or positive H is negative ± exothermic H is positive ± endothermic As a description of changes in heat content and work, these are adequate but they do not describe whether a process is spontaneous or not. There are endothermic processes that are spontaneous ± evaporation of water, the dissolution of ammonium chloride in water, the melting of ice and so on. We need a thermodynamic description of spontaneous processes in order to fully describe a chemical system Chemistry 130 Equilibrium and energy A spontaneous process is one that takes place without any influence external to the system. -

Lecture 13. Thermodynamic Potentials (Ch
Lecture 13. Thermodynamic Potentials (Ch. 5) So far, we have been using the total internal energy U and, sometimes, the enthalpy H to characterize various macroscopic systems. These functions are called the thermodynamic potentials: all the thermodynamic properties of the system can be found by taking partial derivatives of the TP. For each TP, a set of so-called “natural variables” exists: −= + μ NddVPSdTUd = + + μ NddPVSdTHd Today we’ll introduce the other two thermodynamic potentials: theHelmhotzfree energy F and Gibbs free energy G. Depending on the type of a process, one of these four thermodynamic potentials provides the most convenient description (and is tabulated). All four functions have units of energy. When considering different types of Potential Variables processes, we will be interested in two main U (S,V,N) S, V, N issues: H (S,P,N) S, P, N what determines the stability of a system and how the system evolves towards an F (T,V,N) V, T, N equilibrium; G (T,P,N) P, T, N how much work can be extracted from a system. Diffusive Equilibrium and Chemical Potential For completeness, let’s rcall what we’vee learned about the chemical potential. 1 P μ d U T d= S− P+ μ dVd d= S Nd U +dV d − N T T T The meaning of the partial derivative (∂S/∂N)U,V : let’s fix VA and VB (the membrane’s position is fixed), but U , V , S U , V , S A A A B B B assume that the membrane becomes permeable for gas molecules (exchange of both U and N between the sub- ns ilesystems, the molecuA and B are the same ). -

Thermodynamic Potentials and Thermodynamic Relations In
arXiv:1004.0337 Thermodynamic potentials and Thermodynamic Relations in Nonextensive Thermodynamics Guo Lina, Du Jiulin Department of Physics, School of Science, Tianjin University, Tianjin 300072, China Abstract The generalized Gibbs free energy and enthalpy is derived in the framework of nonextensive thermodynamics by using the so-called physical temperature and the physical pressure. Some thermodynamical relations are studied by considering the difference between the physical temperature and the inverse of Lagrange multiplier. The thermodynamical relation between the heat capacities at a constant volume and at a constant pressure is obtained using the generalized thermodynamical potential, which is found to be different from the traditional one in Gibbs thermodynamics. But, the expressions for the heat capacities using the generalized thermodynamical potentials are unchanged. PACS: 05.70.-a; 05.20.-y; 05.90.+m Keywords: Nonextensive thermodynamics; The generalized thermodynamical relations 1. Introduction In the traditional thermodynamics, there are several fundamental thermodynamic potentials, such as internal energy U , Helmholtz free energy F , enthalpy H , Gibbs free energyG . Each of them is a function of temperature T , pressure P and volume V . They and their thermodynamical relations constitute the basis of classical thermodynamics. Recently, nonextensive thermo-statitstics has attracted significent interests and has obtained wide applications to so many interesting fields, such as astrophysics [2, 1 3], real gases [4], plasma [5], nuclear reactions [6] and so on. Especially, one has been studying the problems whether the thermodynamic potentials and their thermo- dynamic relations in nonextensive thermodynamics are the same as those in the classical thermodynamics [7, 8]. In this paper, under the framework of nonextensive thermodynamics, we study the generalized Gibbs free energy Gq in section 2, the heat capacity at constant volume CVq and heat capacity at constant pressure CPq in section 3, and the generalized enthalpy H q in Sec.4. -

Enthalpy and Free Energy of Reaction
KINETICS AND THERMODYNAMICS OF REACTIONS Key words: Enthalpy, entropy, Gibbs free energy, heat of reaction, energy of activation, rate of reaction, rate law, energy profiles, order and molecularity, catalysts INTRODUCTION This module offers a very preliminary detail of basic thermodynamics. Emphasis is given on the commonly used terms such as free energy of a reaction, rate of a reaction, multi-step reactions, intermediates, transition states etc., FIRST LAW OF THERMODYNAMICS. o Law of conservation of energy. States that, • Energy can be neither created nor destroyed. • Total energy of the universe remains constant. • Energy can be converted from one form to another form. eg. Combustion of octane (petrol). 2 C8H18(l) + 25 O2(g) CO2(g) + 18 H2O(g) Conversion of potential energy into thermal energy. → 16 + 10.86 KJ/mol . ESSENTIAL FACTORS FOR REACTION. For a reaction to progress. • The equilibrium must favor the products- • Thermodynamics(energy difference between reactant and product) should be favorable • Reaction rate must be fast enough to notice product formation in a reasonable period. • Kinetics( rate of reaction) ESSENTIAL TERMS OF THERMODYNAMICS. Thermodynamics. Predicts whether the reaction is thermally favorable. The energy difference between the final products and reactants are taken as the guiding principle. The equilibrium will be in favor of products when the product energy is lower. Molecule with lowered energy posses enhanced stability. Essential terms o Free energy change (∆G) – Overall free energy difference between the reactant and the product o Enthalpy (∆H) – Heat content of a system under a given pressure. o Entropy (∆S) – The energy of disorderness, not available for work in a thermodynamic process of a system. -

Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions
CHEM 1B: GENERAL CHEMISTRY Chapter 20: Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions Instructor: Dr. Orlando E. Raola 20-1 Santa Rosa Junior College Chapter 20 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20-2 Thermodynamics: Entropy, Free Energy, and the Direction of Chemical Reactions 20.1 The Second Law of Thermodynamics: Predicting Spontaneous Change 20.2 Calculating Entropy Change of a Reaction 20.3 Entropy, Free Energy, and Work 20.4 Free Energy, Equilibrium, and Reaction Direction 20-3 Spontaneous Change A spontaneous change is one that occurs without a continuous input of energy from outside the system. All chemical processes require energy (activation energy) to take place, but once a spontaneous process has begun, no further input of energy is needed. A nonspontaneous change occurs only if the surroundings continuously supply energy to the system. If a change is spontaneous in one direction, it will be nonspontaneous in the reverse direction. 20-4 The First Law of Thermodynamics Does Not Predict Spontaneous Change Energy is conserved. It is neither created nor destroyed, but is transferred in the form of heat and/or work. DE = q + w The total energy of the universe is constant: DEsys = -DEsurr or DEsys + DEsurr = DEuniv = 0 The law of conservation of energy applies to all changes, and does not allow us to predict the direction of a spontaneous change. 20-5 DH Does Not Predict Spontaneous Change A spontaneous change may be exothermic or endothermic. Spontaneous exothermic processes include: • freezing and condensation at low temperatures, • combustion reactions, • oxidation of iron and other metals. -

Thermodynamics.Pdf
1 Statistical Thermodynamics Professor Dmitry Garanin Thermodynamics February 24, 2021 I. PREFACE The course of Statistical Thermodynamics consist of two parts: Thermodynamics and Statistical Physics. These both branches of physics deal with systems of a large number of particles (atoms, molecules, etc.) at equilibrium. 3 19 One cm of an ideal gas under normal conditions contains NL = 2:69×10 atoms, the so-called Loschmidt number. Although one may describe the motion of the atoms with the help of Newton's equations, direct solution of such a large number of differential equations is impossible. On the other hand, one does not need the too detailed information about the motion of the individual particles, the microscopic behavior of the system. One is rather interested in the macroscopic quantities, such as the pressure P . Pressure in gases is due to the bombardment of the walls of the container by the flying atoms of the contained gas. It does not exist if there are only a few gas molecules. Macroscopic quantities such as pressure arise only in systems of a large number of particles. Both thermodynamics and statistical physics study macroscopic quantities and relations between them. Some macroscopics quantities, such as temperature and entropy, are non-mechanical. Equilibruim, or thermodynamic equilibrium, is the state of the system that is achieved after some time after time-dependent forces acting on the system have been switched off. One can say that the system approaches the equilibrium, if undisturbed. Again, thermodynamic equilibrium arises solely in macroscopic systems. There is no thermodynamic equilibrium in a system of a few particles that are moving according to the Newton's law.