Gibbs Free Energy - Wikipedia, the Free Encyclopedia 頁 1 / 6
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamics Notes
Thermodynamics Notes Steven K. Krueger Department of Atmospheric Sciences, University of Utah August 2020 Contents 1 Introduction 1 1.1 What is thermodynamics? . .1 1.2 The atmosphere . .1 2 The Equation of State 1 2.1 State variables . .1 2.2 Charles' Law and absolute temperature . .2 2.3 Boyle's Law . .3 2.4 Equation of state of an ideal gas . .3 2.5 Mixtures of gases . .4 2.6 Ideal gas law: molecular viewpoint . .6 3 Conservation of Energy 8 3.1 Conservation of energy in mechanics . .8 3.2 Conservation of energy: A system of point masses . .8 3.3 Kinetic energy exchange in molecular collisions . .9 3.4 Working and Heating . .9 4 The Principles of Thermodynamics 11 4.1 Conservation of energy and the first law of thermodynamics . 11 4.1.1 Conservation of energy . 11 4.1.2 The first law of thermodynamics . 11 4.1.3 Work . 12 4.1.4 Energy transferred by heating . 13 4.2 Quantity of energy transferred by heating . 14 4.3 The first law of thermodynamics for an ideal gas . 15 4.4 Applications of the first law . 16 4.4.1 Isothermal process . 16 4.4.2 Isobaric process . 17 4.4.3 Isosteric process . 18 4.5 Adiabatic processes . 18 5 The Thermodynamics of Water Vapor and Moist Air 21 5.1 Thermal properties of water substance . 21 5.2 Equation of state of moist air . 21 5.3 Mixing ratio . 22 5.4 Moisture variables . 22 5.5 Changes of phase and latent heats . -

Thermodynamics of Metal Extraction
THERMODYNAMICS OF METAL EXTRACTION By Rohit Kumar 2019UGMMO21 Ritu Singh 2019UGMM049 Dhiraj Kumar 2019UGMM077 INTRODUCTION In general, a process or extraction of metals has two components: 1. Severance:produces, through chemical reaction, two or more new phases, one of which is much richer in the valuable content than others. 2. Separation : involves physical separation of one phase from the others Three types of processes are used for accomplishing severance : 1. Pyrometallurgical: Chemical reactions take place at high temperatures. 2. Hydrometallurgical: Chemical reactions carried out in aqueous media. 3. Electrometallurgical: Involves electrochemical reactions. Advantages of Pyrometallurgical processes 1. Option of using a cheap reducing agent, (such as C, CO). As a result, the unit cost is 5-10 times lower than that of the reductants used in electrolytic processes where electric power often is the reductant. 2. Enhanced reaction rates at high temperature . 3. Simplified separation of (molten) product phase at high temperature. 4. Simplified recovery of precious metals ( Au,Ag,Cu) at high temperature. 5. Ability to shift reaction equilibria by changing temperatures. THERMODYNAMICS The Gibbs free energy(G) , of the system is the energy available in the system to do work . It is relevant to us here, because at constant temperature and pressure it considers only variables contained within the system . 퐺 = 퐻 − 푇푆 Here, T is the temperature of the system S is the entropy, or disorder, of the system. H is the enthalpy of the system, defined as: 퐻 = 푈 + 푃푉 Where U is the intern energy , P is the pressure and V is the volume. If the system is changed by a small amount , we can differentiate the above functions: 푑퐺 = 푑퐻 − 푇푑푆 − 푆푑푇 And 푑퐻 = 푑푈 − 푃푑푉 + 푉푑푃 From the first law, 푑푈 = 푑푞 − 푑푤 And from second law, 푑푆 = 푑푞/푇 We see that, 푑퐺 = 푑푞 − 푑푤 + 푃푑푉 + 푉푑푃 − 푇푑푆 − 푆푑푇 푑퐺 = −푑푤 + 푃푑푉 + 푉푑푃 − 푆푑푇 (푑푤 = 푃푑푉) 푑퐺 = 푉푑푃 − 푆푑푇 The above equations show that if the temperature and pressure are kept constant, the free energy does not change. -

Lecture 2 the First Law of Thermodynamics (Ch.1)
Lecture 2 The First Law of Thermodynamics (Ch.1) Outline: 1. Internal Energy, Work, Heating 2. Energy Conservation – the First Law 3. Quasi-static processes 4. Enthalpy 5. Heat Capacity Internal Energy The internal energy of a system of particles, U, is the sum of the kinetic energy in the reference frame in which the center of mass is at rest and the potential energy arising from the forces of the particles on each other. system Difference between the total energy and the internal energy? boundary system U = kinetic + potential “environment” B The internal energy is a state function – it depends only on P the values of macroparameters (the state of a system), not on the method of preparation of this state (the “path” in the V macroparameter space is irrelevant). T A In equilibrium [ f (P,V,T)=0 ] : U = U (V, T) U depends on the kinetic energy of particles in a system and an average inter-particle distance (~ V-1/3) – interactions. For an ideal gas (no interactions) : U = U (T) - “pure” kinetic Internal Energy of an Ideal Gas f The internal energy of an ideal gas U = Nk T with f degrees of freedom: 2 B f ⇒ 3 (monatomic), 5 (diatomic), 6 (polyatomic) (here we consider only trans.+rotat. degrees of freedom, and neglect the vibrational ones that can be excited at very high temperatures) How does the internal energy of air in this (not-air-tight) room change with T if the external P = const? f ⎡ PV ⎤ f U =Nin room k= T Bin⎢ N room = ⎥ = PV 2 ⎣ kB T⎦ 2 - does not change at all, an increase of the kinetic energy of individual molecules with T is compensated by a decrease of their number. -
![Arxiv:1806.08325V1 [Quant-Ph] 21 Jun 2018 Between Particle Number and Energy, in the Same Way That Temperature T Acts As an Exchange Rate Between Entropy and Energy](https://docslib.b-cdn.net/cover/4679/arxiv-1806-08325v1-quant-ph-21-jun-2018-between-particle-number-and-energy-in-the-same-way-that-temperature-t-acts-as-an-exchange-rate-between-entropy-and-energy-154679.webp)
Arxiv:1806.08325V1 [Quant-Ph] 21 Jun 2018 Between Particle Number and Energy, in the Same Way That Temperature T Acts As an Exchange Rate Between Entropy and Energy
Quantum Thermodynamics book Quantum thermodynamics with multiple conserved quantities Erick Hinds-Mingo,1 Yelena Guryanova,2 Philippe Faist,3 and David Jennings4, 1 1QOLS, Blackett Laboratory, Imperial College London, London SW7 2AZ, United Kingdom 2Institute for Quantum Optics and Quantum Information (IQOQI), Boltzmanngasse 3 1090, Vienna, Austria 3Institute for Quantum Information and Matter, Caltech, Pasadena CA, 91125 USA 4Department of Physics, University of Oxford, Oxford, OX1 3PU, United Kingdom (Dated: June 22, 2018) In this chapter we address the topic of quantum thermodynamics in the presence of additional observables beyond the energy of the system. In particular we discuss the special role that the generalized Gibbs ensemble plays in this theory, and derive this state from the perspectives of a micro-canonical ensemble, dynamical typicality and a resource-theory formulation. A notable obsta- cle occurs when some of the observables do not commute, and so it is impossible for the observables to simultaneously take on sharp microscopic values. We show how this can be circumvented, discuss information-theoretic aspects of the setting, and explain how thermodynamic costs can be traded between the different observables. Finally, we discuss open problems and future directions for the topic. INTRODUCTION Thermodynamics has been remarkable in its applicability to a vast array of systems. Indeed, the laws of macroscopic thermodynamics have been successfully applied to the studies of magnetization [1, 2], superconductivity [3], cosmology [4], chemical reactions [5] and biological phenomena [6, 7], to name a few fields. In thermodynamics, energy plays a key role as a thermodynamic potential, that is, as a function of the other thermodynamic variables which characterizes all the thermodynamic properties of the system. -

Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump Scott M
Purdue University Purdue e-Pubs CTRC Research Publications Cooling Technologies Research Center 2012 Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump Scott M. Flueckiger Purdue University Fabien Volle Laboratoire des Sciences des Procédés et des Matériaux S V. Garimella Purdue University, [email protected] Rajiv K. Mongia Intel Corporation Follow this and additional works at: http://docs.lib.purdue.edu/coolingpubs Flueckiger, Scott M.; Volle, Fabien; Garimella, S V.; and Mongia, Rajiv K., "Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump" (2012). CTRC Research Publications. Paper 182. http://dx.doi.org/http://dx.doi.org/10.1016/j.enconman.2012.04.015 This document has been made available through Purdue e-Pubs, a service of the Purdue University Libraries. Please contact [email protected] for additional information. Thermodynamic and Kinetic Investigation of a Chemical Reaction-Based Miniature Heat Pump* Scott M. Flueckiger1, Fabien Volle2, Suresh V. Garimella1**, Rajiv K. Mongia3 1 Cooling Technologies Research Center, an NSF I/UCRC School of Mechanical Engineering and Birck Nanotechnology Center 585 Purdue Mall, Purdue University West Lafayette, Indiana 47907-2088 USA 2 Laboratoire des Sciences des Procédés et des Matériaux (LSPM, UPR 3407 CNRS), Université Paris XIII, 99 avenue J. B. Clément, 93430 Villetaneuse, France 3 Intel Corporation Santa Clara, California 95054 USA * Submitted for publication in Energy Conversion and Management ** Author to who correspondence should be addressed: (765) 494-5621, [email protected] Abstract Representative reversible endothermic chemical reactions (paraldehyde depolymerization and 2-proponal dehydrogenation) are theoretically assessed for their use in a chemical heat pump design for compact thermal management applications. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Carbon Dioxide Heat Transfer Coefficients and Pressure
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Archivio della ricerca - Università degli studi di Napoli Federico II international journal of refrigeration 33 (2010) 1068e1085 available at www.sciencedirect.com www.iifiir.org journal homepage: www.elsevier.com/locate/ijrefrig Carbon dioxide heat transfer coefficients and pressure drops during flow boiling: Assessment of predictive methods R. Mastrullo a, A.W. Mauro a, A. Rosato a,*, G.P. Vanoli b a D.E.TE.C., Facolta` di Ingegneria, Universita` degli Studi di Napoli Federico II, p.le Tecchio 80, 80125 Napoli, Italy b Dipartimento di Ingegneria, Universita` degli Studi del Sannio, corso Garibaldi 107, Palazzo dell’Aquila Bosco Lucarelli, 82100 Benevento, Italy article info abstract Article history: Among the alternatives to the HCFCs and HFCs, carbon dioxide emerged as one of the most Received 13 December 2009 promising environmentally friendly refrigerants. In past years many works were carried Received in revised form out about CO2 flow boiling and very different two-phase flow characteristics from 8 February 2010 conventional fluids were found. Accepted 5 April 2010 In order to assess the best predictive methods for the evaluation of CO2 heat transfer Available online 9 April 2010 coefficients and pressure gradients in macro-channels, in the current article a literature survey of works and a collection of the results of statistical comparisons available in Keywords: literature are furnished. Heat exchanger In addition the experimental data from University of Naples are used to run a deeper Boiling analysis. Both a statistical and a direct comparison against some of the most quoted Carbon dioxide-review predictive methods are carried out. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

Thermodynamic Stability: Free Energy and Chemical Equilibrium ©David Ronis Mcgill University
Chemistry 223: Thermodynamic Stability: Free Energy and Chemical Equilibrium ©David Ronis McGill University 1. Spontaneity and Stability Under Various Conditions All the criteria for thermodynamic stability stem from the Clausius inequality,cf. Eq. (8.7.3). In particular,weshowed that for anypossible infinitesimal spontaneous change in nature, d− Q dS ≥ .(1) T Conversely,if d− Q dS < (2) T for every allowed change in state, then the system cannot spontaneously leave the current state NO MATTER WHAT;hence the system is in what is called stable equilibrium. The stability criterion becomes particularly simple if the system is adiabatically insulated from the surroundings. In this case, if all allowed variations lead to a decrease in entropy, then nothing will happen. The system will remain where it is. Said another way,the entropyofan adiabatically insulated stable equilibrium system is a maximum. Notice that the term allowed plays an important role. Forexample, if the system is in a constant volume container,changes in state or variations which lead to a change in the volume need not be considered eveniftheylead to an increase in the entropy. What if the system is not adiabatically insulated from the surroundings? Is there a more convenient test than Eq. (2)? The answer is yes. To see howitcomes about, note we can rewrite the criterion for stable equilibrium by using the first lawas − d Q = dE + Pop dV − µop dN > TdS,(3) which implies that dE + Pop dV − µop dN − TdS >0 (4) for all allowed variations if the system is in equilibrium. Equation (4) is the key stability result. -

New Explicit Correlation for the Compressibility Factor of Natural Gas: Linearized Z-Factor Isotherms
J Petrol Explor Prod Technol (2016) 6:481–492 DOI 10.1007/s13202-015-0209-3 ORIGINAL PAPER - PRODUCTION ENGINEERING New explicit correlation for the compressibility factor of natural gas: linearized z-factor isotherms 1 2 3 Lateef A. Kareem • Tajudeen M. Iwalewa • Muhammad Al-Marhoun Received: 14 October 2014 / Accepted: 17 October 2015 / Published online: 18 December 2015 Ó The Author(s) 2015. This article is published with open access at Springerlink.com Abstract The compressibility factor (z-factor) of gases is Keywords Z-factor Á Explicit correlation Á Reduced a thermodynamic property used to account for the devia- temperature Á Reduced pressure Á Natural gas tion of real gas behavior from that of an ideal gas. Corre- lations based on the equation of state are often implicit, List of symbols because they require iteration and are computationally P Pressure (psi) expensive. A number of explicit correlations have been P Pseudo-critical pressure derived to enhance simplicity; however, no single explicit pc P Pseudo-reduced pressure correlation has been developed for the full range of pseudo- pr ÀÁ T Temperature (R) reduced temperatures 1:05 Tpr 3 and pseudo-reduced ÀÁ Tpc Pseudo-critical temperature (R) pressures 0:2 Ppr 15 , which represents a significant Tpr Pseudo-reduced temperature research gap. This work presents a new z-factor correlation Ppc Pseudo-critical pressure (psi) that can be expressed in linear form. On the basis of Hall Ppr Pseudo-reduced pressure and Yarborough’s implicit correlation, we developed the v Initial guess for iteration process new correlation from 5346 experimental data points Y Pseudo-reduced density extracted from 5940 data points published in the SPE z-factor Compressibility factor natural gas reservoir engineering textbook and created a linear z-factor chart for a full range of pseudo-reduced temperatures ð1:15 Tpr 3Þ and pseudo-reduced pres- sures ð0:2 Ppr 15Þ. -
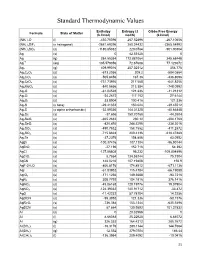
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

New Explicit Correlation for the Compressibility Factor of Natural Gas: Linearized Z-Factor Isotherms
J Petrol Explor Prod Technol DOI 10.1007/s13202-015-0209-3 ORIGINAL PAPER - PRODUCTION ENGINEERING New explicit correlation for the compressibility factor of natural gas: linearized z-factor isotherms 1 2 3 Lateef A. Kareem • Tajudeen M. Iwalewa • Muhammad Al-Marhoun Received: 14 October 2014 / Accepted: 17 October 2015 Ó The Author(s) 2015. This article is published with open access at Springerlink.com Abstract The compressibility factor (z-factor) of gases is Keywords Z-factor Á Explicit correlation Á Reduced a thermodynamic property used to account for the devia- temperature Á Reduced pressure Á Natural gas tion of real gas behavior from that of an ideal gas. Corre- lations based on the equation of state are often implicit, List of symbols because they require iteration and are computationally P Pressure (psi) expensive. A number of explicit correlations have been P Pseudo-critical pressure derived to enhance simplicity; however, no single explicit pc P Pseudo-reduced pressure correlation has been developed for the full range of pseudo- pr ÀÁ T Temperature (R) reduced temperatures 1:05 Tpr 3 and pseudo-reduced ÀÁ Tpc Pseudo-critical temperature (R) pressures 0:2 Ppr 15 , which represents a significant Tpr Pseudo-reduced temperature research gap. This work presents a new z-factor correlation Ppc Pseudo-critical pressure (psi) that can be expressed in linear form. On the basis of Hall Ppr Pseudo-reduced pressure and Yarborough’s implicit correlation, we developed the v Initial guess for iteration process new correlation from 5346 experimental data points Y Pseudo-reduced density extracted from 5940 data points published in the SPE z-factor Compressibility factor natural gas reservoir engineering textbook and created a linear z-factor chart for a full range of pseudo-reduced temperatures ð1:15 Tpr 3Þ and pseudo-reduced pres- sures ð0:2 Ppr 15Þ.