Mechanistic Studies of Some Chemical and Biochemical Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules As Potent Antibacterial and Antifungal Agents
Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules as Potent Antibacterial and Antifungal Agents Dissertation Zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) Vorgelegt der Naturwissenschaftlichen Fakultät I Institut für Pharmazie Fachbereich für Pharmazeutische Chemie der Martin-Luther-Universität Halle-Wittenberg von Kaveh Yasrebi Geboren am 09.14.1987 in Teheran/Iran (Islamische Republik) Gutachter: 1. Prof. Dr. Andreas Hilgeroth (Martin-Luther-Universität Halle-Wittenberg, Germany) 2. Prof. Dr. Sibel Süzen (Ankara Üniversitesi, Turkey) 3. Prof. Dr. Michael Lalk (Ernst-Moritz-Arndt-Universität Greifswald, Germany) Halle (Saale), den 21. Juli 2020 Selbstständigkeitserklärung Hiermit erkläre ich gemäß § 5 (2) b der Promotionsordnung der Naturwissenschaftlichen Fakultät I – Institut für Pharmazie der Martin-Luther-Universität Halle-Wittenberg, dass ich die vorliegende Arbeit selbstständig und ohne Benutzung anderer als der angegebenen Hilfsmittel und Quellen angefertigt habe. Alle Stellen, die wörtlich oder sinngemäß aus Veröffentlichungen entnommen sind, habe ich als solche kenntlich gemacht. Ich erkläre ferner, dass diese Arbeit in gleicher oder ähnlicher Form bisher keiner anderen Prüfbehörde zur Erlangung des Doktorgrades vorgelegt wurde. Halle (Saale), den 21. Juli 2020 Kaveh Yasrebi Acknowledgement This study was carried out from June 2015 to July 2017 in the Research Group of Drug Development and Analysis led by Prof. Dr. Andreas Hilgeroth at the Institute of Pharmacy, Martin-Luther-Universität Halle-Wittenberg. I would like to thank all the people for their participation who supported my work in this way and helped me obtain good results. First of all, I would like to express my gratitude to Prof. Dr. Andreas Hilgeroth for providing me with opportunity to carry out my Ph.D. -

Novel Antimicrobial Agents Inhibiting Lipid II Incorporation Into Peptidoglycan Essay MBB
27 -7-2019 Novel antimicrobial agents inhibiting lipid II incorporation into peptidoglycan Essay MBB Mark Nijland S3265978 Supervisor: Prof. Dr. Dirk-Jan Scheffers Molecular Microbiology University of Groningen Content Abstract..............................................................................................................................................2 1.0 Peptidoglycan biosynthesis of bacteria ........................................................................................3 2.0 Novel antimicrobial agents ...........................................................................................................4 2.1 Teixobactin ...............................................................................................................................4 2.2 tridecaptin A1............................................................................................................................7 2.3 Malacidins ................................................................................................................................8 2.4 Humimycins ..............................................................................................................................9 2.5 LysM ........................................................................................................................................ 10 3.0 Concluding remarks .................................................................................................................... 11 4.0 references ................................................................................................................................. -

Parsek Micro410 Lecture
Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Organization of the Prokaryotic Cell Prokaryotic Structures fimbriae Size Range of Prokaryotes bacillus See Table 4.1 (rigid) vibrio Nanobacteria 0.05‐0.2 µm (0.14‐0.2 µm) (flexible) Thiomargarita namibiensis Mycoplasma are 700‐750 µm (Fig. 4.2) pleomorphic green alga Nanochlorum eukaryotum Mycoplasma 0.1‐ ~1-2 µm in diameter 0.3 µm Fig. 4.1 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Staining cells for Microscopic observation Cell Arrangements Motility- ~80% of prokaryotes are motile streptococcus Staining properties: Gram Stain staphylococcus Fig. 2.3 Gram Stain (1884) (Bacteria) Gram-negative mixed culture Gram-positive Fig. 2.3 and 2.4 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Functions of the cytoplasmic membrane The phospholipid bi‐layer Fig. 4.9 Fig. 4.4 What is the structure of bacterial phospholipids? Other components of the cytoplasmic membrane Figs. 4.5‐4.6 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Archaeal membranes can be a lipid monolayer Archaeal phospholipids have an ether linkage Fig. 4.7 Fig. 4.8 Importance of Cell Wall Schematic diagram cell wall • Provides rigidity to cell allowing cell to withstand the large osmotic/ionic Fig. 4.16 changes a bacterium may experience in its environment, and turgor pressure of cytoplasm (conc. of solutes in cytoplasm). Cell lysis • May have a role in shape determination. • Provides a barrier against certain toxic chemical and biological agents. • Site of action of some of the most commonly used antibiotics used to treat bacterial infections (penicillin family). -
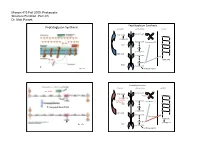
Parsek Lecture #3
Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Peptidoglycan Synthesis Peptidoglycan Synthesis cytoplasm cell membrane cell wall Bactoprenol-P Pi UDP-NAM M G pentapeptide G M Bactoprenol Bactoprenol-P-P P M UMP G P NAM G M pentapeptide M G UDP-NAG Bactoprenol G P NAM‐NAG P NAM-NAG UMP pentapeptide Fig. 6.7a Interbridge peptide Peptidoglycan Synthesis Cross-linking of Peptidoglycan Strands cytoplasm cell membrane cell wall autolysins Bactoprenol-P Pi UDP-NAM Bacitracin M G pentapeptide G D-cycloserine Bactoprenol M (Oxamycin) Bactoprenol-P-P P M UMP G P Transpeptidase (FtsI) NAM G M pentapeptide M G UDP-NAG Bactoprenol G Vancomycin P NAM‐NAG P NAM-NAG pentapeptide UMP Fig. 6.7b pentapeptide Interbridge peptide Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cross-linking of Peptidoglycan Strands Antibiotic Resistance autolysins • Inactivate antibiotic β-lactamase (penicillinase) Clavulanic acid β-lactams Augmentin and Trimentin (combination of clavulanic acid and transpeptidase amoxicillin or ampicillin respectively) penicillins and cephalosporins lysozyme • Change chemistry of target site • Limit access of the antibiotic to target site Fig. 6.5 Cell Shape Determination • Modifications made to Peptidoglycan: ‐ lysozyme: Protoplasts/spheroplasts ‐ autolysins Bacillus subtilis ‐ endopeptidase Heliobacter pylori • Protein(s) may play a major role ‐ MreB protein Caulobacter crescentus ‐ MreB has homology to actin, a component of the cytoskeleton of eukaryotes. Shape determining protein‐ crescentin Fig. 6.4 Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cell Wall Gram-positive Bacteria intermediate filaments in the bacteria Caulobacter crescentus glycerol similar predicted structures of crescentin and intermediate filaments Fig. -

WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C07D 519/00 (2006.01) A61P 39/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C07D 487/04 (2006.01) A61P 35/00 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/5517 (2006.01) A61P 37/00 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, A61K 47/48 (2006.01) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (21) International Application Number: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, PCT/IB2013/058229 SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (22) International Filing Date: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 2 September 2013 (02.09.2013) ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (71) Applicant: HANGZHOU DAC BIOTECH CO., LTD UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, [US/CN]; Room B2001-B2019, Building 2, No 452 Sixth TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Street, Hangzhou Economy Development Area, Hangzhou EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, City, Zhejiang 310018 (CN). -

Antibiotic Discovery
ANTIBIOTIC DISCOVERY RESISTANCE PROFILING OF MICROBIAL GENOMES TO REVEAL NOVEL ANTIBIOTIC NATURAL PRODUCTS By CHELSEA WALKER, H. BSc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfilment of the Requirements for the Degree Master of Science McMaster University © Copyright by Chelsea Walker, May 2017 McMaster University MASTER OF SCIENCE (2017) Hamilton, Ontario (Biochemistry and Biomedical Sciences) TITLE: Resistance Profiling of Microbial Genomes to Reveal Novel Antibiotic Natural Products. AUTHOR: Chelsea Walker, H. BSc. (McMaster University) SUPERVISOR: Dr. Nathan A. Magarvey. COMMITTEE MEMBERS: Dr. Eric Brown and Dr. Michael G. Surette. NUMBER OF PAGES: xvii, 168 ii Lay Abstract It would be hard to imagine a world where we could no longer use the antibiotics we are routinely being prescribed for common bacterial infections. Currently, we are in an era where this thought could become a reality. Although we have been able to discover antibiotics in the past from soil dwelling microbes, this approach to discovery is being constantly challenged. At the same time, the bacteria are getting smarter in their ways to evade antibiotics, in the form of resistance, or self-protection mechanisms. As such is it essential to devise methods which can predict the potential for resistance to the antibiotics we use early in the discovery and isolation process. By using what we have learned in the past about how bacteria protect themselves for antibiotics, we can to stay one step ahead of them as we continue to search for new sources of antibiotics from bacteria. iii Abstract Microbial natural products have been an invaluable resource for providing clinically relevant therapeutics for almost a century, including most of the commonly used antibiotics that are still in medical use today. -

Synthesis and Characterization of Protic Ammonium Based Ionic Liquids
SYNTHESIS AND CHARACTERIZATION OF PROTIC AMMONIUM BASED IONIC LIQUIDS M. Sc THESIS A DISSERTATION SUBMITTED FOR THE PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE (M. Sc) IN ORGANIC CHEMISTRY SUBMITTED BY KHADIZA STUDENT ID: 0416032602F REGISTRATION NO: 0416032602 SESSION: APRIL’ 2016 MAY, 2019 ORGANIC CHEMISTRY RESEARCH LABORATORY DEPARTMENT OF CHEMISTRY BANGLADESH UNIVERSITY OF ENGINEERING AND TECHNOLOGY BUET DHAKA-1000 i Dedicated To ii iii DEPARTMENT OF CHEMISTRY BANGLADESH UNIVERSITY OF ENGINEERING AND TECHNOLOGY BUET DHAKA-1000 STUDENT’S DECLARATION I hereby certify that this material, which I now submit for assessment on the program of study leading to the award of M.Sc is entirely my own work, and I also declare this thesis or any part of it has not been submitted elsewhere for the award of any degree or diploma. Signature of the Student Date: ……………….. .………………...………… KHADIZA (Candidate) ID No. : 0416032602F Session: April‟2016 Department of Chemistry BUET, Dhaka-1000 iv ABSTRACT Ionic Liquids (ILs) have attracted extensive research interest in recent years as environmentally benign solvents and it has also been considered as the “engineering solvents” having its tunable physical and chemical properties. ILs are typically consist of organic nitrogen-containing heterocyclic/aliphatic cations and inorganic or organic anions. The ILs designed and synthesized for carrying out specified task are referred as functionalized ILs, which show great application potential in various processes. The efforts to produce functionalized ILs, characterized them, and evaluate their properties and applications are presented in this thesis. A series of Seven (7) achiral and chiral protic diisopropylethylammonium based ILs were designed, synthesized and characterized. -

E. Coli Pbp1b, Moenomycin-Based
Investigating the Ligand Interactions Between E. coli PBP1b, Moenomycin-based Compounds, and Beta-Lactam Compounds Peter Alexander MSc by Research 2017 i CERTIFICATE OF ORIGINALITY This is to certify that I am responsible for the work submitted in this thesis, that the original work is my own, except as specified in the acknowledgements and in references, and that neither the thesis nor the original work contained therein has been previously submitted to any institution for a degree. Signature: Name: Date: CERTIFICATE OF COMPLIANCE This is to certify that this project has been carried out in accordance with University principles regarding ethics and health and safety. Forms are available to view on request. Signature: Name: Date: ii Abstract Antimicrobial resistance is a growing problem in this era. Resistance to the majority of clinical antibiotics including those of a ‘last line of defence’ nature has been seen in a number of laboratory and clinical settings. One method aiming at reducing this problem is altering existing antimicrobial compounds, in order to improve pharmacological effects (avoiding resistance mechanisms, improved spectrum of use). Analysis of the interactions between the antimicrobial compounds and their targets can determine whether modifications to current antimicrobials (such as moenomycin A, a glycosyltransferase inhibitor) have altered the mode of action. ecoPBP1B is a bifunctional glycosyltransferase that could be used as a model for beta lactams and moenomycins, aiding in the design and development of novel antimicrobials based on these families. Moenomycin A has not seen high clinical usage due to poor pharmacokinetics and bioavailability. This project aimed to show whether ecoPBP1b can be used as a model for novel antimicrobials, such as seeing whether Moenomycin A analogues (with cell penetrating peptides to facilitate entry into the bacterial cell) still retain their ability to bind to glycosyltransferases. -

Lipid II: a Central Component in Bacterial Cell Wall Synthesis and a Target for Antibiotics
ARTICLE IN PRESS Prostaglandins, Leukotrienes and Essential Fatty Acids 79 (2008) 117–121 Contents lists available at ScienceDirect Prostaglandins, Leukotrienes and Essential Fatty Acids journal homepage: www.elsevier.com/locate/plefa Lipid II: A central component in bacterial cell wall synthesis and a target for antibiotics Ben de Kruijff Ã, Vincent van Dam, Eefjan Breukink Chemical Biology and Organic Chemistry, Utrecht University, Padualaan 8, Utrecht, The Netherlands abstract The bacterial cell wall is mainly composed of peptidoglycan, which is a three-dimensional network of long aminosugar strands located on the exterior of the cytoplasmic membrane. These strands consist of alternating MurNAc and GlcNAc units and are interlinked to each other via peptide moieties that are attached to the MurNAc residues. Peptidoglycan subunits are assembled on the cytoplasmic side of the bacterial membrane on a polyisoprenoid anchor and one of the key components in the synthesis of peptidoglycan is Lipid II. Being essential for bacterial cell survival, it forms an attractive target for antibacterial compounds such as vancomycin and several lantibiotics. Lipid II consists of one GlcNAc- MurNAc-pentapeptide subunit linked to a polyiosoprenoid anchor 11 subunits long via a pyrophosphate linker. This review focuses on this special molecule and addresses three questions. First, why are special lipid carriers as polyprenols used in the assembly of peptidoglycan? Secondly, how is Lipid II translocated across the bacterial cytoplasmic membrane? And finally, how is Lipid II used as a receptor for lantibiotics to kill bacteria? & 2008 Elsevier Ltd. All rights reserved. 1. Introduction which will be discussed later. Despite considerable knowledge of cell wall synthesis several key questions remained unanswered so The bacterial cell wall is a unique structure. -

Säurefreie Und Metallfreie Organische Polymere Als Katalysatoren Für Die Umwandlung Von Biomasse
Säurefreie und metallfreie organische Polymere als Katalysatoren für die Umwandlung von Biomasse Dissertation zur Erlangung des akademischen Grades Doktor der Naturwissenschaften (Dr. rer. nat.) Vorgelegt an der Fakultät für Chemie und Biochemie der Ruhr-Universität Bochum von Marc-Philipp Ruby geboren in Magdeburg Bochum Juli 2016 Die vorliegende Arbeit wurde in der Zeit von März 2013 bis Juli 2016 in der Arbeits- gruppe für heterogene Katalyse und funktionelle Feststoffe am Max-Planck-Institut für Kohlenforschung in Mülheim an der Ruhr unter der Leitung von Herrn Prof. Dr. Ferdi Schüth angefertigt. Referent: Prof. Dr. Ferdi Schüth Korreferent: Prof. Dr. Lukas J. Gooßen Tag der Disputation: 02.11.2016 Mit größtem Dank widme ich diese Arbeit meinen Eltern, insbesondere für ihre stetige Unterstützung in jeglicher Form in den letzten Jahren. „Die Wirklichkeit, von der wir sprechen können, ist nie die Wirklichkeit an sich, sondern eine von uns gestaltete Wirklichkeit.“ Werner Karl Heisenberg Danksagung Zuallererst und vor allem gilt mein Dank Professor Dr. Ferdi Schüth für die Ermögli- chung der Durchführung meiner Doktorarbeit und der Überlassung des interessanten und fordernden Themas zur Polymer-katalysierten Umwandlung von Biomasse in seinem Ar- beitskreis. Des Weiteren bin ich besonders dankbar für seine exzellente Betreuung und Unterstützung sowie für die angebotenen lehrreichen Seminare. Darüber hinaus bin ich außerordentlich dankbar über die Freiheit der eigenständigen wissenschaftlichen Bearbei- tung meines Promotionsthemas. Ebenfalls danken möchte ich Professor Dr. Lukas J. Gooßen für das Interesse an mei- nen Projekten und der bereitwilligen Zustimmung zur Erstellung des Zweitgutachtens. Darüber hinaus möchte ich Dr. Felix H. Richter, Valentina Nese und Alessia Padovani für zahlreiche anregende Diskussionen zur Synthese polymerer Katalysatoren und deren Anwendung zur Umwandlung von Biomasse danken. -

Structural Characterization of Bacterial Defense Complex Marko Nedeljković
Structural characterization of bacterial defense complex Marko Nedeljković To cite this version: Marko Nedeljković. Structural characterization of bacterial defense complex. Biomolecules [q-bio.BM]. Université Grenoble Alpes, 2017. English. NNT : 2017GREAV067. tel-03085778 HAL Id: tel-03085778 https://tel.archives-ouvertes.fr/tel-03085778 Submitted on 22 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTE UNIVERSITE GRENOBLE ALPES Spécialité : Biologie Structurale et Nanobiologie Arrêté ministériel : 25 mai 2016 Présentée par Marko NEDELJKOVIĆ Thèse dirigée par Andréa DESSEN préparée au sein du Laboratoire Institut de Biologie Structurale dans l'École Doctorale Chimie et Sciences du Vivant Caractérisation structurale d'un complexe de défense bactérienne Structural characterization of a bacterial defense complex Thèse soutenue publiquement le 21 décembre 2017, devant le jury composé de : Monsieur Herman VAN TILBEURGH Professeur, Université Paris Sud, Rapporteur Monsieur Laurent TERRADOT Directeur de Recherche, Institut de Biologie et Chimie des Protéines, Rapporteur Monsieur Patrice GOUET Professeur, Université Lyon 1, Président Madame Montserrat SOLER-LOPEZ Chargé de Recherche, European Synchrotron Radiation Facility, Examinateur Madame Andréa DESSEN Directeur de Recherche, Institut de Biologie Structurale , Directeur de These 2 Contents ABBREVIATIONS .............................................................................................................................. -

Discovery of Wall Teichoic Acid Inhibitors As Potential Anti-MRSA &Beta
Chemistry & Biology Article Discovery of Wall Teichoic Acid Inhibitors as Potential Anti-MRSA b-Lactam Combination Agents Hao Wang,1 Charles J. Gill,1 Sang H. Lee,1 Paul Mann,1 Paul Zuck,2 Timothy C. Meredith,3 Nicholas Murgolo,1 Xinwei She,1 Susan Kales,1 Lianzhu Liang,1 Jenny Liu,1 Jin Wu,1 John Santa Maria,4 Jing Su,1 Jianping Pan,1 Judy Hailey,1 Debra Mcguinness,1 Christopher M. Tan,1 Amy Flattery,1 Suzanne Walker,4 Todd Black,1 and Terry Roemer1,* 1Infectious Disease Biology, Merck Research Laboratories, Kenilworth, NJ 07033, USA 2Screening and Protein Science, Merck Research Laboratories, West Point, PA 19486, USA 3Infectious Diseases Area, Novartis Institutes for BioMedical Research, Cambridge, MA 02139, USA 4Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, MA 02115, USA *Correspondence: [email protected] http://dx.doi.org/10.1016/j.chembiol.2012.11.013 SUMMARY bacteria (Drawz and Bonomo, 2010). Paramount to the rationale of combination agents is the increased potency and efficacy Innovative strategies are needed to combat drug achieved by their combined effects. Ideally, this is achieved resistance associated with methicillin-resistant by the synergistic bioactivity of both agents affecting two inter- Staphylococcus aureus (MRSA). Here, we investigate dependent cellular processes required for cell growth as well the potential of wall teichoic acid (WTA) biosynthesis as the targeted inactivation of the resistance mechanism to the inhibitors as combination agents to restore b-lactam first agent by the combination agent (Tan et al., 2012). Applying efficacy against MRSA. Performing a whole-cell a systems biology approach to discovering synergistic agents with this therapeutic potential is highly warranted; lethal or pathway-based screen, we identified a series of even growth-crippling chemical genetic interactions highlight WTA inhibitors (WTAIs) targeting the WTA trans- a cellular network of interdependent biological processes and porter protein, TarG.