E. Coli Pbp1b, Moenomycin-Based
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules As Potent Antibacterial and Antifungal Agents
Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules as Potent Antibacterial and Antifungal Agents Dissertation Zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) Vorgelegt der Naturwissenschaftlichen Fakultät I Institut für Pharmazie Fachbereich für Pharmazeutische Chemie der Martin-Luther-Universität Halle-Wittenberg von Kaveh Yasrebi Geboren am 09.14.1987 in Teheran/Iran (Islamische Republik) Gutachter: 1. Prof. Dr. Andreas Hilgeroth (Martin-Luther-Universität Halle-Wittenberg, Germany) 2. Prof. Dr. Sibel Süzen (Ankara Üniversitesi, Turkey) 3. Prof. Dr. Michael Lalk (Ernst-Moritz-Arndt-Universität Greifswald, Germany) Halle (Saale), den 21. Juli 2020 Selbstständigkeitserklärung Hiermit erkläre ich gemäß § 5 (2) b der Promotionsordnung der Naturwissenschaftlichen Fakultät I – Institut für Pharmazie der Martin-Luther-Universität Halle-Wittenberg, dass ich die vorliegende Arbeit selbstständig und ohne Benutzung anderer als der angegebenen Hilfsmittel und Quellen angefertigt habe. Alle Stellen, die wörtlich oder sinngemäß aus Veröffentlichungen entnommen sind, habe ich als solche kenntlich gemacht. Ich erkläre ferner, dass diese Arbeit in gleicher oder ähnlicher Form bisher keiner anderen Prüfbehörde zur Erlangung des Doktorgrades vorgelegt wurde. Halle (Saale), den 21. Juli 2020 Kaveh Yasrebi Acknowledgement This study was carried out from June 2015 to July 2017 in the Research Group of Drug Development and Analysis led by Prof. Dr. Andreas Hilgeroth at the Institute of Pharmacy, Martin-Luther-Universität Halle-Wittenberg. I would like to thank all the people for their participation who supported my work in this way and helped me obtain good results. First of all, I would like to express my gratitude to Prof. Dr. Andreas Hilgeroth for providing me with opportunity to carry out my Ph.D. -

A New Antibiotic Kills Pathogens Without Detectable Resistance
ARTICLE doi:10.1038/nature14098 A new antibiotic kills pathogens without detectable resistance Losee L. Ling1*, Tanja Schneider2,3*, Aaron J. Peoples1, Amy L. Spoering1, Ina Engels2,3, Brian P. Conlon4, Anna Mueller2,3, Till F. Scha¨berle3,5, Dallas E. Hughes1, Slava Epstein6, Michael Jones7, Linos Lazarides7, Victoria A. Steadman7, Douglas R. Cohen1, Cintia R. Felix1, K. Ashley Fetterman1, William P. Millett1, Anthony G. Nitti1, Ashley M. Zullo1, Chao Chen4 & Kim Lewis4 Antibiotic resistance is spreading faster than the introduction of new compounds into clinical practice, causing a public health crisis. Most antibiotics were produced by screening soil microorganisms, but this limited resource of cultivable bacteria was overmined by the 1960s. Synthetic approaches to produce antibiotics have been unable to replace this platform. Uncultured bacteria make up approximately 99% of all species in external environments, and are an untapped source of new antibiotics. We developed several methods to grow uncultured organisms by cultivation in situ or by using specific growth factors. Here we report a new antibiotic that we term teixobactin, discovered in a screen of uncultured bacteria. Teixobactin inhibits cell wall synthesis by binding to a highly conserved motif of lipid II (precursor of peptidoglycan) and lipid III (precursor of cell wall teichoic acid). We did not obtain any mutants of Staphylococcus aureus or Mycobacterium tuberculosis resistant to teixobactin. The properties of this compound suggest a path towards developing antibiotics that are likely to avoid development of resistance. Widespread introduction of antibiotics in the 1940s, beginning with factors through the chambers enables growth of uncultured bacteria in penicillin1,2 and streptomycin3, transformed medicine, providing effec- their natural environment. -

Novel Antimicrobial Agents Inhibiting Lipid II Incorporation Into Peptidoglycan Essay MBB
27 -7-2019 Novel antimicrobial agents inhibiting lipid II incorporation into peptidoglycan Essay MBB Mark Nijland S3265978 Supervisor: Prof. Dr. Dirk-Jan Scheffers Molecular Microbiology University of Groningen Content Abstract..............................................................................................................................................2 1.0 Peptidoglycan biosynthesis of bacteria ........................................................................................3 2.0 Novel antimicrobial agents ...........................................................................................................4 2.1 Teixobactin ...............................................................................................................................4 2.2 tridecaptin A1............................................................................................................................7 2.3 Malacidins ................................................................................................................................8 2.4 Humimycins ..............................................................................................................................9 2.5 LysM ........................................................................................................................................ 10 3.0 Concluding remarks .................................................................................................................... 11 4.0 references ................................................................................................................................. -

Parsek Micro410 Lecture
Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Organization of the Prokaryotic Cell Prokaryotic Structures fimbriae Size Range of Prokaryotes bacillus See Table 4.1 (rigid) vibrio Nanobacteria 0.05‐0.2 µm (0.14‐0.2 µm) (flexible) Thiomargarita namibiensis Mycoplasma are 700‐750 µm (Fig. 4.2) pleomorphic green alga Nanochlorum eukaryotum Mycoplasma 0.1‐ ~1-2 µm in diameter 0.3 µm Fig. 4.1 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Staining cells for Microscopic observation Cell Arrangements Motility- ~80% of prokaryotes are motile streptococcus Staining properties: Gram Stain staphylococcus Fig. 2.3 Gram Stain (1884) (Bacteria) Gram-negative mixed culture Gram-positive Fig. 2.3 and 2.4 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Functions of the cytoplasmic membrane The phospholipid bi‐layer Fig. 4.9 Fig. 4.4 What is the structure of bacterial phospholipids? Other components of the cytoplasmic membrane Figs. 4.5‐4.6 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Archaeal membranes can be a lipid monolayer Archaeal phospholipids have an ether linkage Fig. 4.7 Fig. 4.8 Importance of Cell Wall Schematic diagram cell wall • Provides rigidity to cell allowing cell to withstand the large osmotic/ionic Fig. 4.16 changes a bacterium may experience in its environment, and turgor pressure of cytoplasm (conc. of solutes in cytoplasm). Cell lysis • May have a role in shape determination. • Provides a barrier against certain toxic chemical and biological agents. • Site of action of some of the most commonly used antibiotics used to treat bacterial infections (penicillin family). -
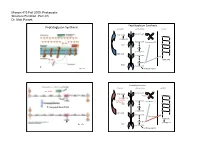
Parsek Lecture #3
Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Peptidoglycan Synthesis Peptidoglycan Synthesis cytoplasm cell membrane cell wall Bactoprenol-P Pi UDP-NAM M G pentapeptide G M Bactoprenol Bactoprenol-P-P P M UMP G P NAM G M pentapeptide M G UDP-NAG Bactoprenol G P NAM‐NAG P NAM-NAG UMP pentapeptide Fig. 6.7a Interbridge peptide Peptidoglycan Synthesis Cross-linking of Peptidoglycan Strands cytoplasm cell membrane cell wall autolysins Bactoprenol-P Pi UDP-NAM Bacitracin M G pentapeptide G D-cycloserine Bactoprenol M (Oxamycin) Bactoprenol-P-P P M UMP G P Transpeptidase (FtsI) NAM G M pentapeptide M G UDP-NAG Bactoprenol G Vancomycin P NAM‐NAG P NAM-NAG pentapeptide UMP Fig. 6.7b pentapeptide Interbridge peptide Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cross-linking of Peptidoglycan Strands Antibiotic Resistance autolysins • Inactivate antibiotic β-lactamase (penicillinase) Clavulanic acid β-lactams Augmentin and Trimentin (combination of clavulanic acid and transpeptidase amoxicillin or ampicillin respectively) penicillins and cephalosporins lysozyme • Change chemistry of target site • Limit access of the antibiotic to target site Fig. 6.5 Cell Shape Determination • Modifications made to Peptidoglycan: ‐ lysozyme: Protoplasts/spheroplasts ‐ autolysins Bacillus subtilis ‐ endopeptidase Heliobacter pylori • Protein(s) may play a major role ‐ MreB protein Caulobacter crescentus ‐ MreB has homology to actin, a component of the cytoskeleton of eukaryotes. Shape determining protein‐ crescentin Fig. 6.4 Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cell Wall Gram-positive Bacteria intermediate filaments in the bacteria Caulobacter crescentus glycerol similar predicted structures of crescentin and intermediate filaments Fig. -

WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C07D 519/00 (2006.01) A61P 39/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C07D 487/04 (2006.01) A61P 35/00 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/5517 (2006.01) A61P 37/00 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, A61K 47/48 (2006.01) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (21) International Application Number: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, PCT/IB2013/058229 SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (22) International Filing Date: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 2 September 2013 (02.09.2013) ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (71) Applicant: HANGZHOU DAC BIOTECH CO., LTD UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, [US/CN]; Room B2001-B2019, Building 2, No 452 Sixth TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Street, Hangzhou Economy Development Area, Hangzhou EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, City, Zhejiang 310018 (CN). -

Escherichia Coli K–12 Derived from the Ecocyc Database Daniel S Weaver1*,Ingridmkeseler1,Amandamackie2, Ian T Paulsen2 and Peter D Karp1
Weaver et al. BMC Systems Biology 2014, 8:79 http://www.biomedcentral.com/1752-0509/8/79 RESEARCH ARTICLE OpenAccess A genome-scale metabolic flux model of Escherichia coli K–12 derived from the EcoCyc database Daniel S Weaver1*,IngridMKeseler1,AmandaMackie2, Ian T Paulsen2 and Peter D Karp1 Abstract Background: Constraint-based models of Escherichia coli metabolic flux have played a key role in computational studies of cellular metabolism at the genome scale. We sought to develop a next-generation constraint-based E. coli model that achieved improved phenotypic prediction accuracy while being frequently updated and easy to use. We also sought to compare model predictions with experimental data to highlight open questions in E. coli biology. Results: We present EcoCyc–18.0–GEM, a genome-scale model of the E. coli K–12 MG1655 metabolic network. The model is automatically generated from the current state of EcoCyc using the MetaFlux software, enabling the release of multiple model updates per year. EcoCyc–18.0–GEM encompasses 1445 genes, 2286 unique metabolic reactions, and 1453 unique metabolites. We demonstrate a three-part validation of the model that breaks new ground in breadth and accuracy: (i) Comparison of simulated growth in aerobic and anaerobic glucose culture with experimental results from chemostat culture and simulation results from the E. coli modeling literature. (ii) Essentiality prediction for the 1445 genes represented in the model, in which EcoCyc–18.0–GEM achieves an improved accuracy of 95.2% in predicting the growth phenotype of experimental gene knockouts. (iii) Nutrient utilization predictions under 431 different media conditions, for which the model achieves an overall accuracy of 80.7%. -

Antibiotic Discovery
ANTIBIOTIC DISCOVERY RESISTANCE PROFILING OF MICROBIAL GENOMES TO REVEAL NOVEL ANTIBIOTIC NATURAL PRODUCTS By CHELSEA WALKER, H. BSc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfilment of the Requirements for the Degree Master of Science McMaster University © Copyright by Chelsea Walker, May 2017 McMaster University MASTER OF SCIENCE (2017) Hamilton, Ontario (Biochemistry and Biomedical Sciences) TITLE: Resistance Profiling of Microbial Genomes to Reveal Novel Antibiotic Natural Products. AUTHOR: Chelsea Walker, H. BSc. (McMaster University) SUPERVISOR: Dr. Nathan A. Magarvey. COMMITTEE MEMBERS: Dr. Eric Brown and Dr. Michael G. Surette. NUMBER OF PAGES: xvii, 168 ii Lay Abstract It would be hard to imagine a world where we could no longer use the antibiotics we are routinely being prescribed for common bacterial infections. Currently, we are in an era where this thought could become a reality. Although we have been able to discover antibiotics in the past from soil dwelling microbes, this approach to discovery is being constantly challenged. At the same time, the bacteria are getting smarter in their ways to evade antibiotics, in the form of resistance, or self-protection mechanisms. As such is it essential to devise methods which can predict the potential for resistance to the antibiotics we use early in the discovery and isolation process. By using what we have learned in the past about how bacteria protect themselves for antibiotics, we can to stay one step ahead of them as we continue to search for new sources of antibiotics from bacteria. iii Abstract Microbial natural products have been an invaluable resource for providing clinically relevant therapeutics for almost a century, including most of the commonly used antibiotics that are still in medical use today. -

Synthesis and Structure−Activity Relationships of Teixobactin
Ann. N.Y. Acad. Sci. ISSN 0077-8923 ANNALS OF THE NEW YORK ACADEMY OF SCIENCES Special Issue: Antimicrobial Therapeutics Reviews REVIEW Synthesis and structure−activity relationships of teixobactin John A. Karas,1 Fan Chen,1 Elena K. Schneider-Futschik,1,2 Zhisen Kang,1 Maytham Hussein,1 James Swarbrick,1 Daniel Hoyer,1,3,4 Andrew M. Giltrap,5 Richard J. Payne,5 Jian Li,6 and Tony Velkov1 1Department of Pharmacology & Therapeutics, School of Biomedical Sciences, Faculty of Medicine, Dentistry and Health Sciences, the University of Melbourne, Parkville, Victoria, Australia. 2Lung Health Research Centre, Department of Pharmacology & Therapeutics, the University of Melbourne, Parkville, Victoria, Australia. 3The Florey Institute of Neuroscience and Mental Health, the University of Melbourne, Parkville, Victoria, Australia. 4Department of Molecular Medicine, the Scripps Research Institute, La Jolla, California. 5School of Chemistry, the University of Sydney, Sydney, New South Wales, Australia. 6Monash Biomedicine Discovery Institute, Department of Microbiology, Monash University, Clayton, Victoria, Australia Address for correspondence: Tony Velkov and John A. Karas, Department of Pharmacology & Therapeutics, School of Biomedical Sciences, Faculty of Medicine, Dentistry and Health Sciences, the University of Melbourne, Parkville, VIC 3010, Australia. [email protected] OR [email protected] The discovery of antibiotics has led to the effective treatment of bacterial infections that were otherwise fatal and has had a transformative effect on modern medicine. Teixobactin is an unusual depsipeptide natural product that was recently discovered from a previously unculturable soil bacterium and found to possess potent antibacterial activity against several Gram positive pathogens, including methicillin-resistant Staphylococcus aureus and vancomycin- resistant Enterococci. -

Lipid II As a Target for Antibiotics
Nature Reviews Drug Discovery | AOP, published online 10 March 2006; doi:10.1038/nrd2004 REVIEWS Lipid II as a target for antibiotics Eefjan Breukink and Ben de Kruijff Abstract | Lipid II is a membrane-anchored cell-wall precursor that is essential for bacterial cell-wall biosynthesis. The effectiveness of targeting Lipid II as an antibacterial strategy is highlighted by the fact that it is the target for at least four different classes of antibiotic, including the clinically important glycopeptide antibiotic vancomycin. However, the growing problem of bacterial resistance to many current drugs, including vancomycin, has led to increasing interest in the therapeutic potential of other classes of compound that target Lipid II. Here, we review progress in understanding of the antibacterial activities of these compounds, which include lantibiotics, mannopeptimycins and ramoplanin, and consider factors that will be important in exploiting their potential as new treatments for bacterial infections. Since the discovery of penicillin more than 75 years ago, for novel antibacterial drugs, and here we review their antibiotics have had an immense impact on the treatment mode of action and their antibacterial activities, and use of infections caused by bacteria. However, the widespread, this as a basis to discuss their potential as novel drugs for and sometimes inappropriate, use of antibiotics has gen- combating antibiotic-resistant bacteria. erated a strong evolutionary pressure for the emergence of bacteria that either have an inherent resistance to a The role of Lipid II in cell-wall synthesis particular antibiotic or have the capacity to acquire such The cell wall (FIG. 1) of all bacteria comprises a polymer of resistance. -

1 Molecular Dynamics Simulations Reveal the Conformational Flexibility of Lipid II and Its Loose Association with the Defensin
Molecular dynamics simulations reveal the conformational flexibility of Lipid II and its loose association with the defensin plectasin in the Staphylococcus aureus membrane. Sarah Witzke,1,2 , Michael Petersen1, Timothy S. Carpenter3*and Syma Khalid2* 1 Department of Physics, Chemistry and Pharmacy University of Southern Denmark Denmark 2 School of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. 3 Biosciences and Biotechnology Division, Lawrence Livermore National Laboratory, 7000 East Avenue, Livermore, CA, USA. *To whom correspondence should be addressed: [email protected] or [email protected] 1 Abbreviations and Textual Footnotes ADPG Tetra-anteiso-myristoyl Cardiolipin Ala Alanine ALPG Lysyl-AMPG AMPG 1,2-di-anteiso-myristoyl-sn-glycero-3-phosphoglycerol Cys Cysteine DMPG 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol DPC Dodecylphosphocholine Glc N-acetylglucosamine Gly Glycine His Histidine LII Lipid II Lys Lysine MD Molecular Dynamics Mur N-acetylmuramic acid PG Phosphatidylglycerol Phe Phenylalanine RDF Radial Distribution Function S. aureus Staphylococcus aureus SI Supporting Information 2 Abstract Lipid II is a precursor for peptidoglycan, which is the main component of the bacterial cell wall. Lipid II is a relatively conserved and important part of the cell wall biosynthesis pathway and is targeted by antibiotics such as the lantibiotics, which achieve their function by disrupting the biosynthesis of the cell wall. Given the urgent need for development of novel antibiotics to counter the growing threat of bacterial infection, it is imperative that a thorough molecular-level characterisation of the molecules targeted by antibiotics is achieved. To this end, we present a molecular dynamics simulation study of the conformational dynamics of Lipid II within a detailed model of the Staphylococcus aureus cell membrane. -

As the Peptidoglycan Lipid II Flippase in Escherichia Coli
Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli Natividad Ruiz* Department of Molecular Biology, Princeton University, Princeton, NJ 08544 Communicated by Thomas J. Silhavy, Princeton University, Princeton, NJ, August 25, 2008 (received for review August 7, 2008) Peptidoglycan is a cell-wall glycopeptide polymer that protects in a process that is not well understood but that is known to bacteria from osmotic lysis. Whereas in Gram-positive bacteria it involve dephosphorylation by multiple phosphatases and trans- also serves as scaffold for many virulence factors, in Gram-negative port across the IM (1, 12). In E. coli, recycled and newly bacteria, peptidoglycan is an anchor for the outer membrane. For synthesized undecaprenyl phosphate can be used in new rounds years, we have known the enzymes required for the biosynthesis of peptidoglycan biosynthesis as well as in the transport across of peptidoglycan; what was missing was the flippase that trans- the IM of other cell envelope polysaccharides, such as lipopoly- locates the lipid-anchored precursors across the cytoplasmic mem- saccharides (LPS) and enterobacterial common antigen (ECA) brane before their polymerization into mature peptidoglycan. (13, 14). Thus, although we do not understand how undecaprenyl Using a reductionist bioinformatics approach, I have identified the pyrophosphate is flipped back across the IM for recycling, the essential inner-membrane protein MviN (renamed MurJ) as a likely only unknown factor required for an essential step unique to candidate for the peptidoglycan flippase in Escherichia coli. Here, peptidoglycan biogenesis is the flippase that transports lipid II I present genetic and biochemical data that confirm the require- across the IM.