Staphylococcus Aureus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Interview with Pierre Pichot
PSYCHOPHARMACOLOGY AND THE HISTORY OF PSYCHIATRY PIERRE PICHOT Could we begin with your recollections of the 1955 Paris meeting, which was effectively the first world wide meeting on chlorpromazine. The meeting was organised in Paris by Jean Delay. It was supported by Specia, the pharmaceutical firm which had produced chlorpromazine, which was a branch of the Rhone Poulenc Group. For the first time people engaged in what was called psychopharmacology came together. They came from many countries, including the United States. The efficacy of the drug and the mechanism of action were discussed. However, at that time the biochemistry of the brain, as it exists now, was unknown. It was only at the beginning of the 60s, that we began to speak of the role of the neurotransmitters in the action of both neuroleptic drugs and antidepressants and of their potential abnormalities in the disease process. So in practice 1955 was only a meeting on therapy with chlorpromazine. I have always been very much impressed by the fact that chlorpromazine, which had been introduced only 3 years before, was already used all over the world. Theoretical ideas take usually a very long time to travel from one country to another, and sometimes they never make it. I quote always, the case of Karl Jaspers’ General Psychopathology, which is considered in the German speaking world as one of the basic books of psychiatry. This was published in 1913 but appeared in English translation only in 1963, 50 years later and, even then, a paper published in the American Journal of Psychiatry wrote ingenuously that, until this publication, many psychiatrists in the United States had not realised that Jaspers was not only a philosopher, but also a psychiatrist. -

Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules As Potent Antibacterial and Antifungal Agents
Synthesis and Biological Evaluation of Trisindolyl-Cycloalkanes and Bis- Indolyl Naphthalene Small Molecules as Potent Antibacterial and Antifungal Agents Dissertation Zur Erlangung des akademischen Grades doctor rerum naturalium (Dr. rer. nat.) Vorgelegt der Naturwissenschaftlichen Fakultät I Institut für Pharmazie Fachbereich für Pharmazeutische Chemie der Martin-Luther-Universität Halle-Wittenberg von Kaveh Yasrebi Geboren am 09.14.1987 in Teheran/Iran (Islamische Republik) Gutachter: 1. Prof. Dr. Andreas Hilgeroth (Martin-Luther-Universität Halle-Wittenberg, Germany) 2. Prof. Dr. Sibel Süzen (Ankara Üniversitesi, Turkey) 3. Prof. Dr. Michael Lalk (Ernst-Moritz-Arndt-Universität Greifswald, Germany) Halle (Saale), den 21. Juli 2020 Selbstständigkeitserklärung Hiermit erkläre ich gemäß § 5 (2) b der Promotionsordnung der Naturwissenschaftlichen Fakultät I – Institut für Pharmazie der Martin-Luther-Universität Halle-Wittenberg, dass ich die vorliegende Arbeit selbstständig und ohne Benutzung anderer als der angegebenen Hilfsmittel und Quellen angefertigt habe. Alle Stellen, die wörtlich oder sinngemäß aus Veröffentlichungen entnommen sind, habe ich als solche kenntlich gemacht. Ich erkläre ferner, dass diese Arbeit in gleicher oder ähnlicher Form bisher keiner anderen Prüfbehörde zur Erlangung des Doktorgrades vorgelegt wurde. Halle (Saale), den 21. Juli 2020 Kaveh Yasrebi Acknowledgement This study was carried out from June 2015 to July 2017 in the Research Group of Drug Development and Analysis led by Prof. Dr. Andreas Hilgeroth at the Institute of Pharmacy, Martin-Luther-Universität Halle-Wittenberg. I would like to thank all the people for their participation who supported my work in this way and helped me obtain good results. First of all, I would like to express my gratitude to Prof. Dr. Andreas Hilgeroth for providing me with opportunity to carry out my Ph.D. -

Novel Antimicrobial Agents Inhibiting Lipid II Incorporation Into Peptidoglycan Essay MBB
27 -7-2019 Novel antimicrobial agents inhibiting lipid II incorporation into peptidoglycan Essay MBB Mark Nijland S3265978 Supervisor: Prof. Dr. Dirk-Jan Scheffers Molecular Microbiology University of Groningen Content Abstract..............................................................................................................................................2 1.0 Peptidoglycan biosynthesis of bacteria ........................................................................................3 2.0 Novel antimicrobial agents ...........................................................................................................4 2.1 Teixobactin ...............................................................................................................................4 2.2 tridecaptin A1............................................................................................................................7 2.3 Malacidins ................................................................................................................................8 2.4 Humimycins ..............................................................................................................................9 2.5 LysM ........................................................................................................................................ 10 3.0 Concluding remarks .................................................................................................................... 11 4.0 references ................................................................................................................................. -

Assessing Neurotoxicity of Drugs of Abuse
National Institute on Drug Abuse RESEARCH MONOGRAPH SERIES Assessing Neurotoxicity of Drugs of Abuse 136 U.S. Department of Health and Human Services • Public Health Service • National Institutes of Health Assessing Neurotoxicity of Drugs of Abuse Editor: Lynda Erinoff, Ph.D. NIDA Research Monograph 136 1993 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service National Institutes of Health National Institute on Drug Abuse 5600 Fishers Lane Rockville, MD 20857 ACKNOWLEDGMENT This monograph is based on the papers and discussions from a technical review on “Assessing Neurotoxicity of Drugs of Abuse” held on May 20-21, 1991, in Bethesda, MD. The technical review was sponsored by the National Institute on Drug Abuse (NIDA). COPYRIGHT STATUS NIDA has obtained permission from the copyright holders to reproduce certain previously published material as noted in the text. Further reproduction of this copyrighted material is permitted only as part of a reprinting of the entire publication or chapter. For any other use, the copyright holder’s permission is required. All other material in this volume except quoted passages from copyrighted sources is in the public domain and may be used or reproduced without permission from the Institute or the authors. Citation of the source is appreciated. Opinions expressed in this volume are those of the authors and do not necessarily reflect the opinions or official policy of the National Institute on Drug Abuse or any other part of the U.S. Department of Health and Human Services. The U.S. Government does not endorse or favor any specific commercial product or company. -

Parsek Micro410 Lecture
Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Organization of the Prokaryotic Cell Prokaryotic Structures fimbriae Size Range of Prokaryotes bacillus See Table 4.1 (rigid) vibrio Nanobacteria 0.05‐0.2 µm (0.14‐0.2 µm) (flexible) Thiomargarita namibiensis Mycoplasma are 700‐750 µm (Fig. 4.2) pleomorphic green alga Nanochlorum eukaryotum Mycoplasma 0.1‐ ~1-2 µm in diameter 0.3 µm Fig. 4.1 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Staining cells for Microscopic observation Cell Arrangements Motility- ~80% of prokaryotes are motile streptococcus Staining properties: Gram Stain staphylococcus Fig. 2.3 Gram Stain (1884) (Bacteria) Gram-negative mixed culture Gram-positive Fig. 2.3 and 2.4 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Functions of the cytoplasmic membrane The phospholipid bi‐layer Fig. 4.9 Fig. 4.4 What is the structure of bacterial phospholipids? Other components of the cytoplasmic membrane Figs. 4.5‐4.6 Microm 410 Fall 2009: Prokaryotic Structure/Function part 1 Dr. Matt Parsek Archaeal membranes can be a lipid monolayer Archaeal phospholipids have an ether linkage Fig. 4.7 Fig. 4.8 Importance of Cell Wall Schematic diagram cell wall • Provides rigidity to cell allowing cell to withstand the large osmotic/ionic Fig. 4.16 changes a bacterium may experience in its environment, and turgor pressure of cytoplasm (conc. of solutes in cytoplasm). Cell lysis • May have a role in shape determination. • Provides a barrier against certain toxic chemical and biological agents. • Site of action of some of the most commonly used antibiotics used to treat bacterial infections (penicillin family). -
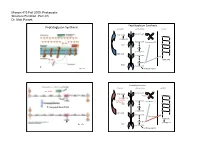
Parsek Lecture #3
Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Peptidoglycan Synthesis Peptidoglycan Synthesis cytoplasm cell membrane cell wall Bactoprenol-P Pi UDP-NAM M G pentapeptide G M Bactoprenol Bactoprenol-P-P P M UMP G P NAM G M pentapeptide M G UDP-NAG Bactoprenol G P NAM‐NAG P NAM-NAG UMP pentapeptide Fig. 6.7a Interbridge peptide Peptidoglycan Synthesis Cross-linking of Peptidoglycan Strands cytoplasm cell membrane cell wall autolysins Bactoprenol-P Pi UDP-NAM Bacitracin M G pentapeptide G D-cycloserine Bactoprenol M (Oxamycin) Bactoprenol-P-P P M UMP G P Transpeptidase (FtsI) NAM G M pentapeptide M G UDP-NAG Bactoprenol G Vancomycin P NAM‐NAG P NAM-NAG pentapeptide UMP Fig. 6.7b pentapeptide Interbridge peptide Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cross-linking of Peptidoglycan Strands Antibiotic Resistance autolysins • Inactivate antibiotic β-lactamase (penicillinase) Clavulanic acid β-lactams Augmentin and Trimentin (combination of clavulanic acid and transpeptidase amoxicillin or ampicillin respectively) penicillins and cephalosporins lysozyme • Change chemistry of target site • Limit access of the antibiotic to target site Fig. 6.5 Cell Shape Determination • Modifications made to Peptidoglycan: ‐ lysozyme: Protoplasts/spheroplasts ‐ autolysins Bacillus subtilis ‐ endopeptidase Heliobacter pylori • Protein(s) may play a major role ‐ MreB protein Caulobacter crescentus ‐ MreB has homology to actin, a component of the cytoskeleton of eukaryotes. Shape determining protein‐ crescentin Fig. 6.4 Microm 410 Fall 2009: Prokaryotic Structure/Function: Part 2/3 Dr. Matt Parsek Cell Wall Gram-positive Bacteria intermediate filaments in the bacteria Caulobacter crescentus glycerol similar predicted structures of crescentin and intermediate filaments Fig. -

Characterization of Multiple Sites of Action of Ibogaine
——Chapter 6—— CHARACTERIZATION OF MULTIPLE SITES OF ACTION OF IBOGAINE Henry Sershen, Audrey Hashim, And Abel Lajtha Nathan Kline Institute Orangeburg, New York 10962 I. Introduction.................................................................................................................. II. Issues Related to Ibogaine in the Treatment of Drug Dependence............................. A. Dopamine as a Primary Site of Drug-Mediated Responses .................................. B. Ibogaine or Its Metabolite and Acute versus Long-Term Effect........................... C. Single or Multiple Sites of Action of Ibogaine ..................................................... III. Effect of Ibogaine on Drug-Induced Behavior............................................................ IV. Binding Site Activity ................................................................................................... A. Relevant Site of Action.......................................................................................... V. Functional Activity ...................................................................................................... VI. Stimulant Drug Actions/Behaviors.............................................................................. VII. Current Non-Ibogaine Drug Treatment Protocols ....................................................... VIII. Conclusions.................................................................................................................. References................................................................................................................... -

WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/028850 Al 5 March 2015 (05.03.2015) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C07D 519/00 (2006.01) A61P 39/00 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, C07D 487/04 (2006.01) A61P 35/00 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, A61K 31/5517 (2006.01) A61P 37/00 (2006.01) HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KN, KP, KR, A61K 47/48 (2006.01) KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, (21) International Application Number: OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, PCT/IB2013/058229 SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, (22) International Filing Date: TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, 2 September 2013 (02.09.2013) ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, SZ, TZ, (71) Applicant: HANGZHOU DAC BIOTECH CO., LTD UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, [US/CN]; Room B2001-B2019, Building 2, No 452 Sixth TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, Street, Hangzhou Economy Development Area, Hangzhou EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, LV, City, Zhejiang 310018 (CN). -

Antibiotic Discovery
ANTIBIOTIC DISCOVERY RESISTANCE PROFILING OF MICROBIAL GENOMES TO REVEAL NOVEL ANTIBIOTIC NATURAL PRODUCTS By CHELSEA WALKER, H. BSc. A Thesis Submitted to the School of Graduate Studies in Partial Fulfilment of the Requirements for the Degree Master of Science McMaster University © Copyright by Chelsea Walker, May 2017 McMaster University MASTER OF SCIENCE (2017) Hamilton, Ontario (Biochemistry and Biomedical Sciences) TITLE: Resistance Profiling of Microbial Genomes to Reveal Novel Antibiotic Natural Products. AUTHOR: Chelsea Walker, H. BSc. (McMaster University) SUPERVISOR: Dr. Nathan A. Magarvey. COMMITTEE MEMBERS: Dr. Eric Brown and Dr. Michael G. Surette. NUMBER OF PAGES: xvii, 168 ii Lay Abstract It would be hard to imagine a world where we could no longer use the antibiotics we are routinely being prescribed for common bacterial infections. Currently, we are in an era where this thought could become a reality. Although we have been able to discover antibiotics in the past from soil dwelling microbes, this approach to discovery is being constantly challenged. At the same time, the bacteria are getting smarter in their ways to evade antibiotics, in the form of resistance, or self-protection mechanisms. As such is it essential to devise methods which can predict the potential for resistance to the antibiotics we use early in the discovery and isolation process. By using what we have learned in the past about how bacteria protect themselves for antibiotics, we can to stay one step ahead of them as we continue to search for new sources of antibiotics from bacteria. iii Abstract Microbial natural products have been an invaluable resource for providing clinically relevant therapeutics for almost a century, including most of the commonly used antibiotics that are still in medical use today. -

Anew Drug Design Strategy in the Liht of Molecular Hybridization Concept
www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 “Drug Design strategy and chemical process maximization in the light of Molecular Hybridization Concept.” Subhasis Basu, Ph D Registration No: VB 1198 of 2018-2019. Department Of Chemistry, Visva-Bharati University A Draft Thesis is submitted for the partial fulfilment of PhD in Chemistry Thesis/Degree proceeding. DECLARATION I Certify that a. The Work contained in this thesis is original and has been done by me under the guidance of my supervisor. b. The work has not been submitted to any other Institute for any degree or diploma. c. I have followed the guidelines provided by the Institute in preparing the thesis. d. I have conformed to the norms and guidelines given in the Ethical Code of Conduct of the Institute. e. Whenever I have used materials (data, theoretical analysis, figures and text) from other sources, I have given due credit to them by citing them in the text of the thesis and giving their details in the references. Further, I have taken permission from the copyright owners of the sources, whenever necessary. IJCRT2012039 International Journal of Creative Research Thoughts (IJCRT) www.ijcrt.org 284 www.ijcrt.org © 2020 IJCRT | Volume 8, Issue 12 December 2020 | ISSN: 2320-2882 f. Whenever I have quoted written materials from other sources I have put them under quotation marks and given due credit to the sources by citing them and giving required details in the references. (Subhasis Basu) ACKNOWLEDGEMENT This preface is to extend an appreciation to all those individuals who with their generous co- operation guided us in every aspect to make this design and drawing successful. -

E. Coli Pbp1b, Moenomycin-Based
Investigating the Ligand Interactions Between E. coli PBP1b, Moenomycin-based Compounds, and Beta-Lactam Compounds Peter Alexander MSc by Research 2017 i CERTIFICATE OF ORIGINALITY This is to certify that I am responsible for the work submitted in this thesis, that the original work is my own, except as specified in the acknowledgements and in references, and that neither the thesis nor the original work contained therein has been previously submitted to any institution for a degree. Signature: Name: Date: CERTIFICATE OF COMPLIANCE This is to certify that this project has been carried out in accordance with University principles regarding ethics and health and safety. Forms are available to view on request. Signature: Name: Date: ii Abstract Antimicrobial resistance is a growing problem in this era. Resistance to the majority of clinical antibiotics including those of a ‘last line of defence’ nature has been seen in a number of laboratory and clinical settings. One method aiming at reducing this problem is altering existing antimicrobial compounds, in order to improve pharmacological effects (avoiding resistance mechanisms, improved spectrum of use). Analysis of the interactions between the antimicrobial compounds and their targets can determine whether modifications to current antimicrobials (such as moenomycin A, a glycosyltransferase inhibitor) have altered the mode of action. ecoPBP1B is a bifunctional glycosyltransferase that could be used as a model for beta lactams and moenomycins, aiding in the design and development of novel antimicrobials based on these families. Moenomycin A has not seen high clinical usage due to poor pharmacokinetics and bioavailability. This project aimed to show whether ecoPBP1b can be used as a model for novel antimicrobials, such as seeing whether Moenomycin A analogues (with cell penetrating peptides to facilitate entry into the bacterial cell) still retain their ability to bind to glycosyltransferases. -

Influence of Antipsychotic and Anticholinergic Loads on Cognitive Functions in Patients with Schizophrenia
Hindawi Publishing Corporation Schizophrenia Research and Treatment Volume 2016, Article ID 8213165, 10 pages http://dx.doi.org/10.1155/2016/8213165 Research Article Influence of Antipsychotic and Anticholinergic Loads on Cognitive Functions in Patients with Schizophrenia Michael Rehse,1,2 Marina Bartolovic,1 Katlehn Baum,3 Dagmar Richter,1 Matthias Weisbrod,1,3 and Daniela Roesch-Ely1 1 Department of Psychiatry, University of Heidelberg, 69115 Heidelberg, Germany 2Psychiatrisches Zentrum Nordbaden, 69168 Wiesloch, Germany 3Department of Psychiatry and Psychotherapy, SRH Hospital Karlsbad-Langensteinbach, 76307 Karlsbad, Germany Correspondence should be addressed to Daniela Roesch-Ely; daniela [email protected] Received 14 October 2015; Revised 19 February 2016; Accepted 7 March 2016 AcademicEditor:L.Citrome Copyright © 2016 Michael Rehse et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Many patients with schizophrenia show cognitive impairment. There is evidence that, beyond a certain dose of antipsychotic medication, the antipsychotic daily dose (ADD) may impair cognitive performance. Parallel to their D2 receptor antagonism, many antipsychotics show a significant binding affinity to cholinergic muscarinic receptors. Pharmacological treatment with a high anticholinergic daily dose (CDD) significantly impairs attention and memory performance. To examine the relationships between individual cognitive performance and ADD and/or CDD, we conducted a retrospective record-based analysis of a sample of = 104 in patients with a diagnosis of schizophrenia, all of whom had completed a comprehensive neuropsychological test battery. To calculate the individual ADD and CDD, the medication at the time of testing was converted according to equivalence models.